

가족성 삼출성 유리체 망막병증(Familial Exudative Vitreoretinopathy, FEVR)은 비정상적인 망막 혈관 발달을 특징으로 하는, 불완전한 말초 망막 혈관 형성과 그에 따른 망막 허혈(retinal ischemia)을 유발하는 희귀 유전성 망막 질환입니다. FEVR의 무혈관 망막은 저산소증(hypoxia)을 유발하고 유리체로 확장되는 신생 혈관의 성장을 자극하여 궁극적으로 유리체 망막 견인(vitreoretinal traction), 망막하 삼출(subretinal exudation), 황반하 출혈(hemorrhage beneath the macula), 망막 주름(retinal fold), 견인성 망막 박리(tractional retinal detachment), 황반 변위(macular displacement) 등의 말기 합병증을 유발합니다.
FEVR의 임상 양상은 비대칭성을 보이는 경우가 많으며 동일한 가족 내 구성원 간에 현저하게 다를 수 있습니다. 경증의 경우 무증상일 수 있지만 중증의 경우 심각한 시력 손실을 동반할 수 있습니다. 현재, FEVR을 치료하는 특정 약물은 없지만 레이저 요법 및 유리체 절제술과 같은 치료는 증상을 완화하고 시력을 보존하는 데 도움이 될 수 있습니다[1].
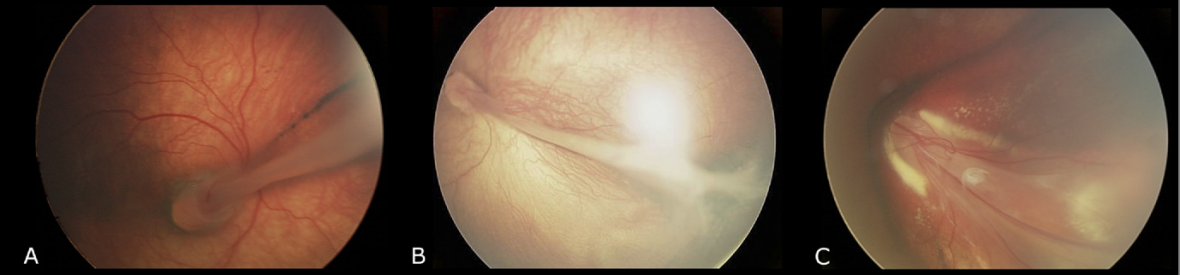
그림 1. 가족성 삼출성 유리체 망막병증(Familial Exudative Vitreoretinopathy, FEVR)의 망막 주름 표현형[2].
혈관 발달은 다양한 신호 전달 경로에 의해 복잡하게 조절되며 그 중 Wnt 신호 전달 경로는 주요 조절 시스템 중 하나입니다. 노린/β-카테닌(Norrin/β-Catenin) 신호 전달 경로는 눈과 귀의 혈관 발달에 중추적인 역할을 하는 Wnt 신호 전달 계통의 중요한 분지입니다[3]. 이 경로에서, 주로 노린(Norrin)과 그 수용체(FZD4, LRP5, TSPAN12) 사이의 복합체 형성을 통해 조절이 일어나며, 이를 통해 망막 혈관 형성을 촉진합니다. 가족성 삼출성 유리체 망막병증(Familial Exudative Vitreoretinopathy, FEVR)을 유발하는 것으로 알려진 유전자 돌연변이는 NDP, FZD4, LRP5, TSPAN12, ZNF408, KIF11, RCBTB1, CTNNB1, JAG1 등 9가지 이상이며, 이는 전 세계 전체 FEVR 사례의 약 50%를 차지하고 있습니다. 이러한 유전적 돌연변이는 Wnt 신호 전달 경로의 정상적인 조절을 방해하여 비정상적인 망막 혈관 발달과 망막 허혈(retinal ischemia) 등의 증상을 야기합니다[4].

그림 2. 망막 혈관 발달에서의 노린/β-카테닌(Norrin/β-Catenin) 신호 조절 경로[4].
NDP 유전자는 망막 혈관계의 성장 및 발달과 관련된 노린(Norrin) 단백질을 부호화합니다. 노린(Norrin) 단백질은 FZD4 단백질과 결합하여 고친화성 리간드-수용체 쌍을 형성합니다. 이 단백질은 보조 구성 요소인 TSPAN12와 함께, β-카테닌(β-Catenin)의 핵 내 진입을 촉진하여 기존 Wnt 경로의 FZD4 및 LRP5 의존적 활성화를 유도합니다. Wnt 신호 전달 경로는 안구의 성장과 발달에 중요한 역할을 하며, 이 경로의 결함은 이 과정에 영향을 미치고 FEVR 및 노리병(Norrie disease)의 발병에 중요한 역할을 담당할 수 있습니다[5].
따라서, NDP 유전자의 돌연변이는 노린(Norrin) 단백질의 기능에 영향을 미치게 되어 결과적으로 이는 Wnt 신호 전달 경로에 영향을 미치고 FEVR의 발병을 야기할 수 있습니다. 마우스(mouse)의 경우, NDP 유전자를 녹아웃시키면 표면 망막 혈관의 발달이 지연되고 심부 망막 혈관이 형성되지 않으며 전형적인 망막 혈관 결함 표현형을 반영하는 미세동맥류(microaneurysm)와 유사한 병변이 형성됩니다[6].
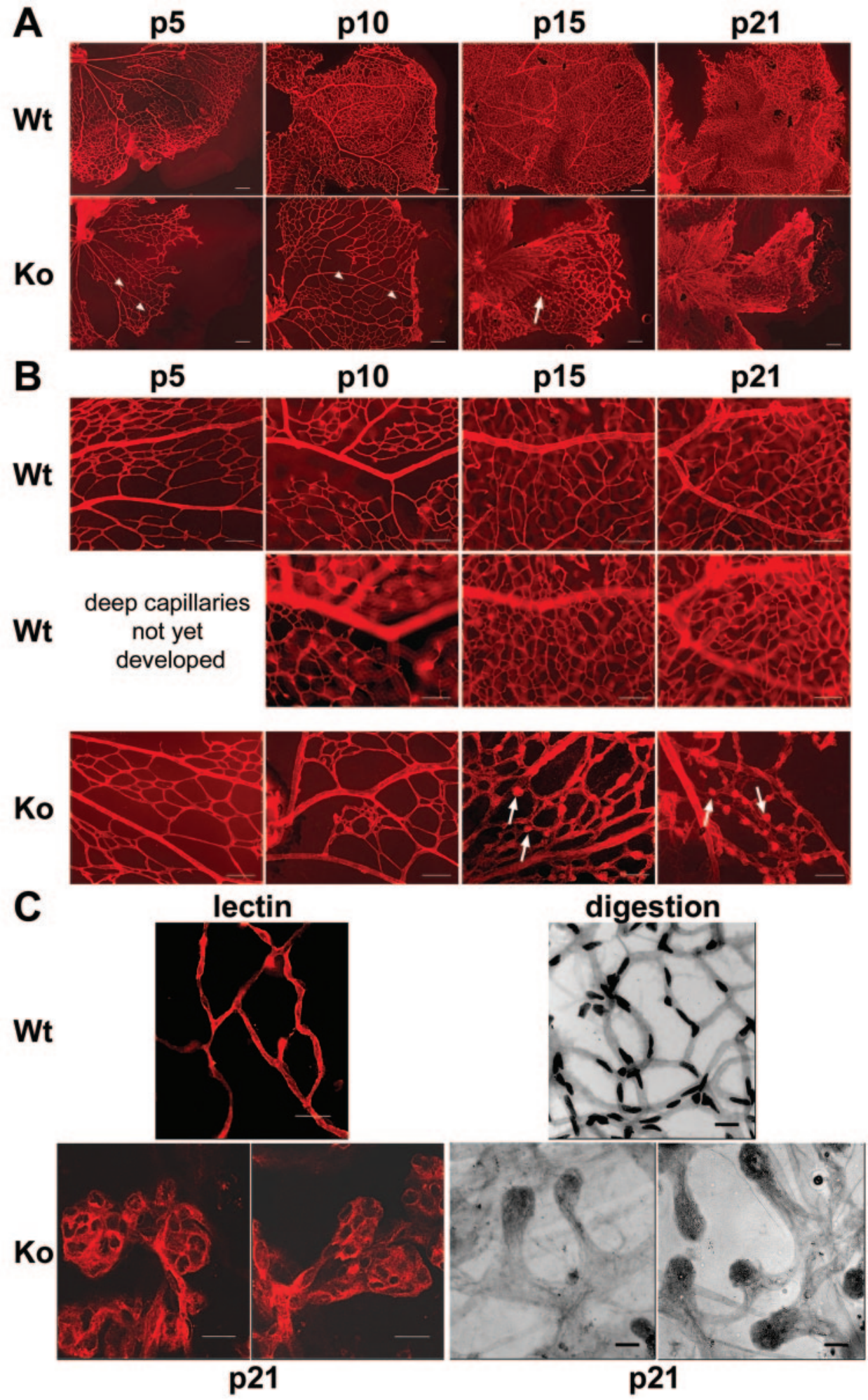
그림 3. NDP-KO 마우스의 표면 망막 혈관계의 결함 및 심부 모세혈관망의 결손[6].
FZD4는 Wnt 신호 전달 경로의 수용체로서, 노린(Norrin)과의 상호작용을 통해 Wnt 단백질에 결합하는 프리즐드-4(Frizzled-4) 단백질을 부호화하여 이 경로를 활성화함으로써 정상적인 망막 혈관 발달을 촉진합니다. FZD4 유전자에 돌연변이가 발생하면, 이 유전자가 Wnt 단백질과 정상적으로 결합하지 못하여 Wnt 신호 전달 경로가 억제됩니다. 이러한 이상은 정맥동맥류(venous aneurysm)와 망막 허혈(retinal ischemia) 증상을 유발할 수 있습니다. 이에 더해, FZD4 유전자의 돌연변이는 TGF-β 및 노치(Notch) 경로와 같은 다른 신호 전달 경로의 조절에도 영향을 미쳐 망막 혈관 발달 및 유지에 영향을 미칠 수 있습니다. 이 두 가지 기전은 함께 작동하여 FEVR의 발생 또는 악화에 영향을 미칠 수 있습니다[7].
NDP 녹아웃(KO) 마우스에서 관찰된 표현형과 유사하게, 동형 접합 FZD4 KO 마우스(mouse)(-/-)는 망막과 내이의 혈관 발달에 상당한 영향을 미치며 망막 스트레스 표현형을 보입니다. FZD4가 결손된 경우 망막 표면 내피 세포의 이동 지연, 망막의 이차 및 삼차 혈관 분지의 제거, 유리체 혈관계의 예정된 퇴행의 현저한 지연, 달팽이관의 점진적인 확장 및 퇴행, 소뇌 혈관의 점진적인 파괴 등 다양한 표현형 결과가 발생합니다. 연구에 따르면 FZD4 결손은 추가적으로 뇌의 퇴화 및 청각 장애와 같은 비혈관성 표현형도 유발할 수 있음을 시사합니다[8].
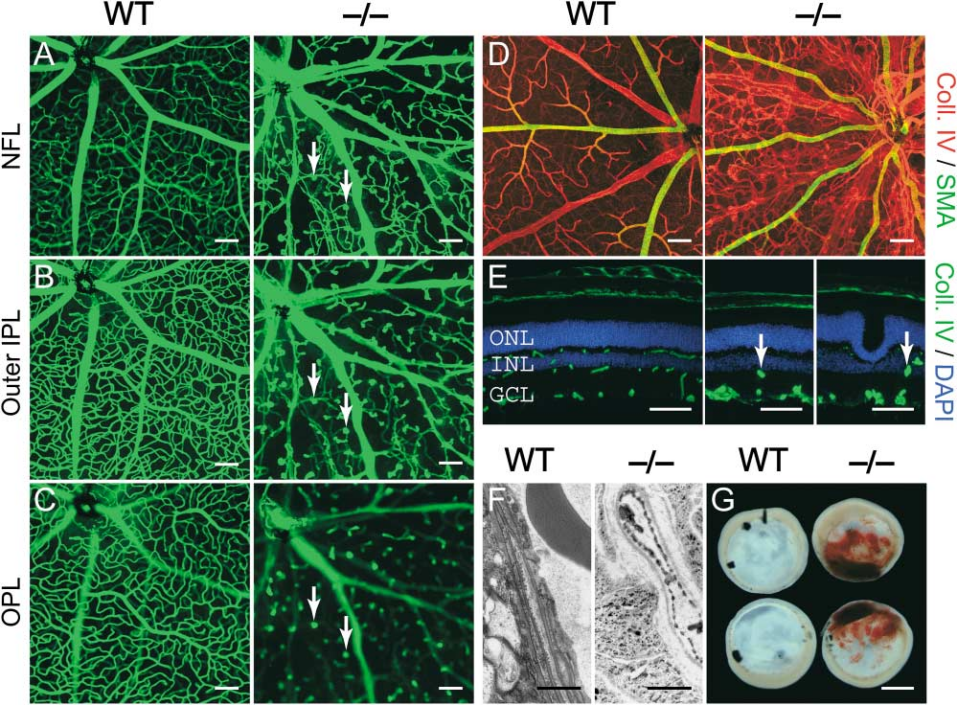
그림 4. 동형 접합 FZD4-KO 마우스(-/-)에서의 망막 혈관 결함 표현형[8].
TSPAN12 유전자에 의해 부호화되는 막관통 단백질(transmembrane protein)인 테트라스패닌-12(Tetraspanin-12)도 Wnt 신호 전달 경로의 중요한 구성 요소입니다. TSPAN12는 FZD4와 상호작용함으로써 Wnt 신호 전달 경로의 조절에 기여하여 정상적인 망막 혈관 발달을 촉진합니다. TSPAN12 유전자의 돌연변이는 FZD4 돌연변이로 인한 것과 유사한 표현형을 야기하며 이 둘 모두 정맥동맥류(venous aneurysm)의 형성 및 망막 허혈(retinal ischemia) 증상을 유발합니다. 이러한 돌연변이는 VEGF 및 노치(Notch)와 같은 신호 전달 경로에 미치는 영향을 통해 망막 혈관 발달 및 유지에 영향을 미칩니다[9].
이와 유사하게, TSPAN12 유전자 녹아웃 마우스는 정맥동맥류(venous aneurysm)의 형성, 비정상적인 망막 혈관의 분지(branching), 혈관 퇴행(vessel regression) 등 Fzd4 녹아웃 마우스와 유사한 표현형을 보입니다. 이에 더해, 마우스의 TSPAN12 결손은 시각 장애, 망막 허혈(retinal ischemia)은 물론 심장 이상, 간 이상, 골격 기형 등 다른 장기 및 조직의 발달 이상을 유발합니다[10].
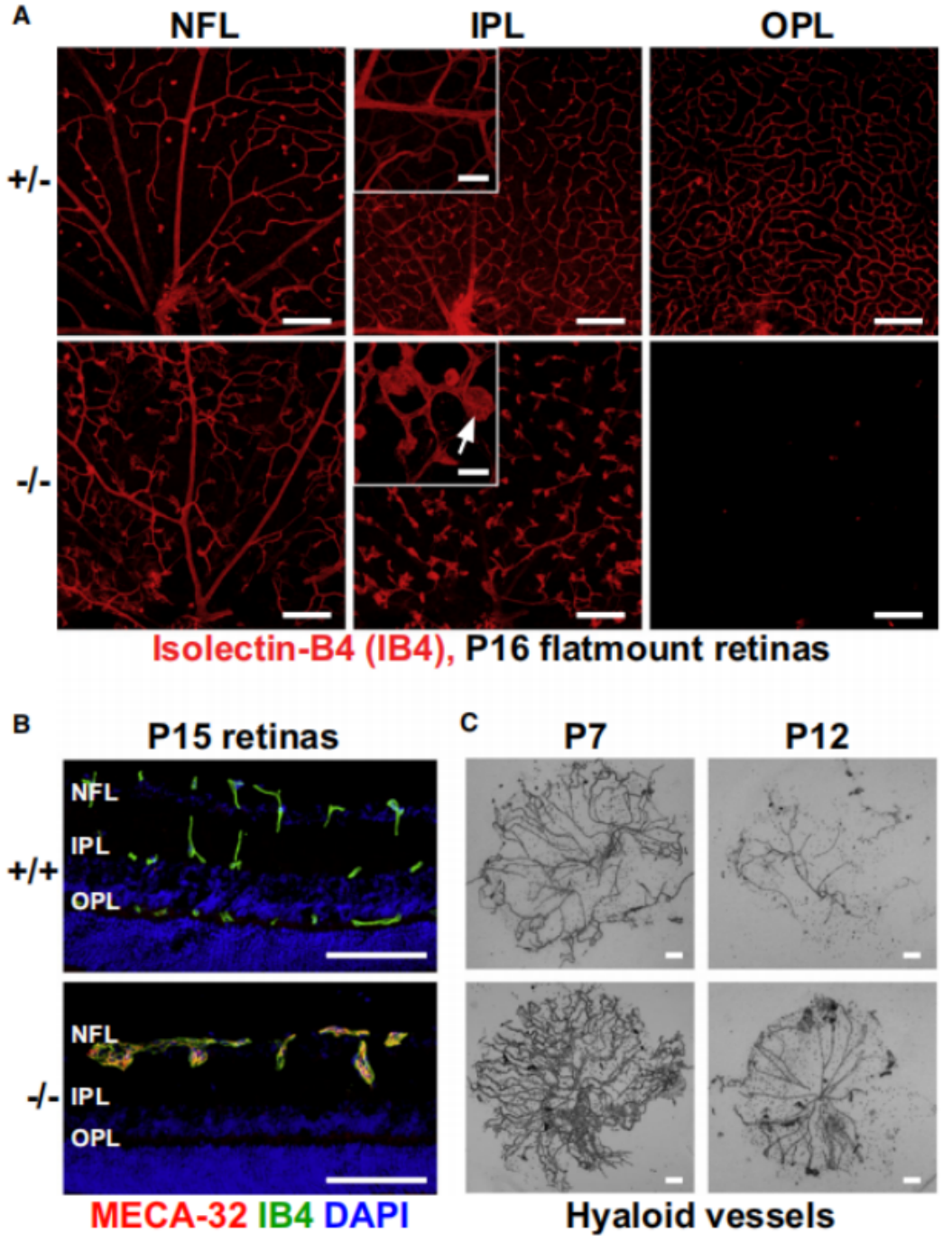
그림 5. Tspan12-KO 마우스에서의 미세동맥류(microaneurysm)의 표현형과 유리체 혈관계의 퇴행 지연[10].
LRP5 유전자는 막관통 단백질(transmembrane protein)(저밀도 지질단백질 수용체 관련 단백질 5(Low-density lipoprotein receptor-related protein 5))을 부호화하며 이 단백질은 Wnt 신호 전달 경로에 관여하는 LRP5/LRP6/Frizzled 공동 수용체 그룹의 핵심 구성 요소로 작용합니다. LRP5는 Wnt 단백질에 결합하여 노린/β-카테닌(Norrin/β-Catenin) 신호 전달 경로를 활성화하여 망막 혈관 형성 조절에 영향을 미칩니다. LRP5 유전자의 기능 소실(loss-of-function) 돌연변이는 Wnt 단백질과의 결합을 방해하여 Wnt 신호 전달 경로 이상을 야기하고 이는 정맥동맥류(venous aneurysm) 형성 및 망막 허혈(retinal ischemia) 증상 등 비정상적인 망막 혈관 발달로 이어져 궁극적으로 FEVR을 유발합니다. 그 외에, LRP5는 골밀도와 콜레스테롤 대사의 조절에도 관여합니다. LRP5 유전자의 기능 소실(loss-of-function) 돌연변이는 골다공증-가성신경교종 증후군(osteoporosis-pseudoglioma syndrome, OPPG)을 유발할 수도 있습니다. 이와 대조적으로, LRP5의 기능 획득(gain-of-function) 돌연변이는 비정상적으로 골밀도를 증가시킬 수 있습니다[11]. 따라서, LRP5 유전자 녹아웃(KO) 마우스는 망막 혈관 이상, 망막 발달 불량 등의 안과 질환 표현형 뿐만 아니라 골격 기형, 낮은 골밀도 등의 골격 표현형과 콜레스테롤 대사 이상, 고콜레스테롤혈증 등의 심혈관 표현형도 보입니다[12-14]. NDP 및 FZD4 유전자 KO 마우스와 비교해, LRP5-KO 마우스는 보다 경증의 망막 병변을 보이며 골격 기형 및 대사 이상 등의 비망막 혈관 표현형을 보입니다.

그림 6. LRP5-KO 마우스의 결함 유리체 혈관 퇴행[12].
CTNNB1 유전자는 카드헤린(cadherin) 및 α-카테닌(α-catenin)과 함께 부착 연접(adherens junction) 복합체를 형성하는 세포질 부착 단백질인 β-카테닌(β-catenin) 단백질을 부호화합니다. 이러한 복합체는 세포 성장과 부착을 조절하여 상피세포층의 형성 및 유지에 중요한 역할을 담당합니다. 일반적으로, CTNNB1 유전자의 손실은 중증 신경 발달 장애인 CTNNB1 증후군을 유발할 수 있는 것으로 알려져 있습니다. 최근 연구에 따르면, CTNNB1 유전자의 특정 기능 소실(loss-of-function) 돌연변이가 NEDSDV(경련성 마비(spastic diplegia) 및 시각 결함을 동반한 신경 발달 장애) 및 가족성 삼출성 유리체 망막병증(Familial Exudative Vitreoretinopathy, FEVR) 등의 망막 결함과 관련된 질환을 유발할 수 있는 것으로 나타났습니다[15-16].
마우스를 대상으로 한 연구에 따르면, CTNNB1 유전자의 전신(동형 접합) 녹아웃(KO)이 배아 사망률, 배아 발달 이상, 신경관 결손, 간 발달 이상, 신장 발달 이상, 중증 심장 발달 표현형을 유발하는 것으로 나타났지만, 망막 표현형에 대한 보고는 제한적입니다. 최근 연구에 따르면, 내피 세포 특이적 CTNNB1의 녹아웃은 노린/β-카테닌(Norrin/β-catenin) 경로의 활성을 감소시켜 망막 혈관 발달에 영향을 미치는 것으로 밝혀졌습니다. 이 마우스 모델에서는 망막 혈관 발달이 억제되어 FEVR 유사 표현형을 보였습니다[17]. 이에 더해, α-카테닌(α-catenin)을 부호화하는 내피 세포 특이적 CTNNB1 유전자 결손은 CTNNB1 결손 마우스와 유사한 FEVR 유사 표현형을 보여 조직 특이적 모델에서 이 유전 질환을 연구할 수 있는 또 다른 방법을 제공합니다[18].
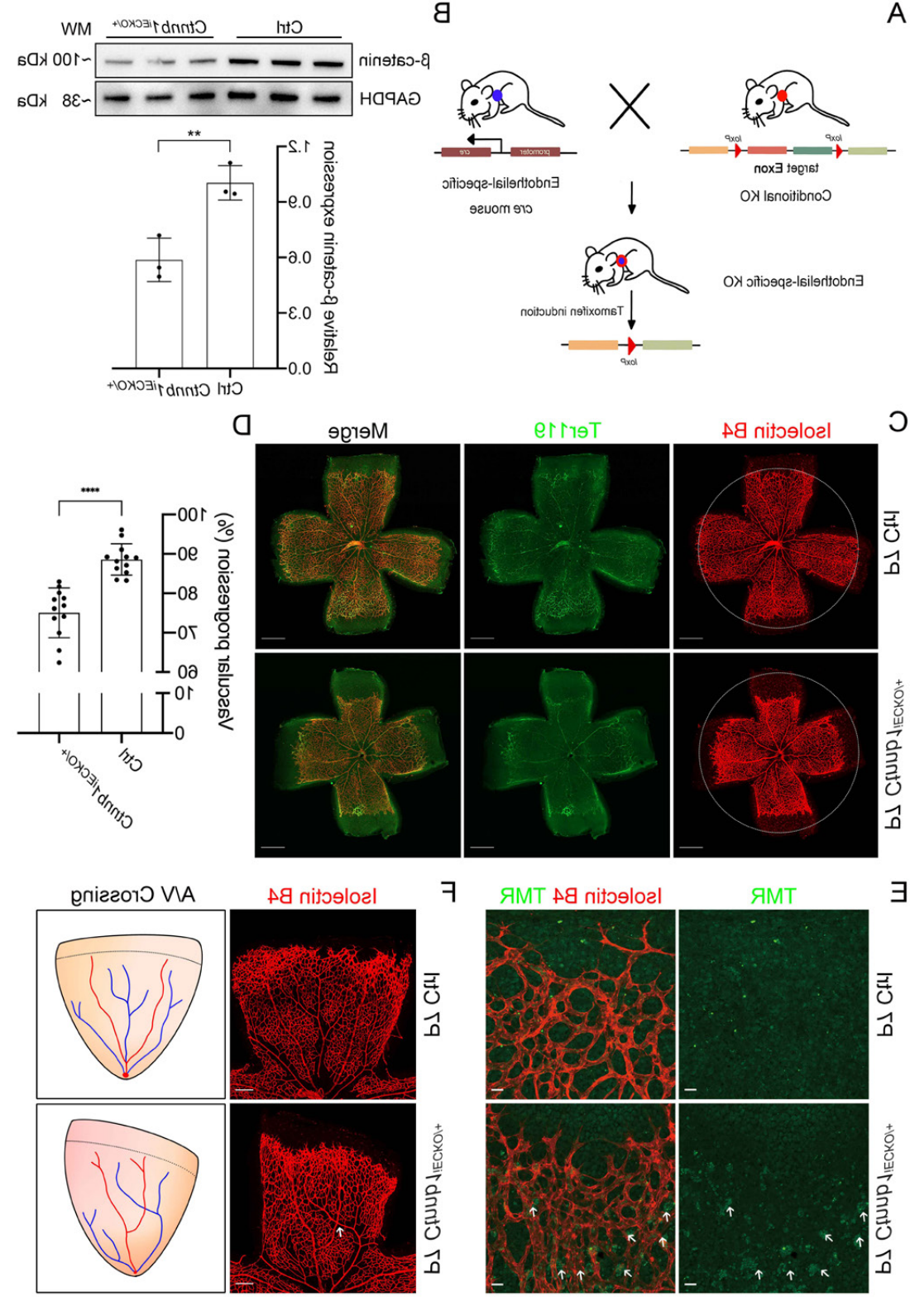
그림 7. 내피 세포 특이적 CTNNB1 녹아웃 마우스는 FEVR 유사 표현형을 보입니다[17].
KIF11은 노린/β-카테닌(Norrin/β-Catenin) 신호 전달 경로에 영향을 받지 않는, FEVR의 또 다른 중요한 원인 유전자입니다. 이 유전자는 세포 분열과 미세소관 역학 등 다양한 생물학적 과정에 관여하는 운동 단백질(키네신 패밀리 멤버 11(Kinesin Family Member 11))을 부호화합니다. 또한, KIF11은 망막 전구 세포(retinal progenitor cell)의 중요한 인자로서 미세소관 역학을 조절하고 망막 혈관의 성장과 분지에도 관여합니다. KIF11의 비활성화 돌연변이는 인간에서, 소두증(microcephaly), 림프부종(lymphedema), 맥락막 망막 이형성증(choroidal retinal dysplasia) 및/또는 지적 장애를 동반하거나 동반하지 않는 망막 혈관 저형성증(retinal vascular hypoplasia)의 다양한 조합을 특징으로 하는 다기관 증후군과 관련되어 있습니다. 여러 연구에 따르면, 일부 FEVR 환자에서 KIF11의 돌연변이가 망막 혈관의 발달과 안정성에 영향을 미칠 수 있지만 그 정확한 기전은 아직 밝혀지지 않았습니다. 이에 따라, KIF11 돌연변이와 FEVR 간의 관계를 연구하는 데 KIF11 유전자 녹아웃 마우스 모델이 사용되었습니다.
최근 연구는 내피 세포 특이적 Kif11 녹아웃(KO)은 마우스의 망막 혈관계에서는 중증의 발달 지연을 유발하며 소뇌 혈관계에서는 경증의 발달 지연을 유발함을 시사합니다[19]. 내피 세포 특이적 Kif11-cKO(conditional KO, 조건부 KO) 마우스의 표현형은 내피 세포 특이적 CTNNB1-cKO 마우스의 표현형과 매우 유사하여 KIF11의 소실과 FEVR 간의 밀접한 연관성을 추가적으로 뒷받침합니다.

그림 8. 초기 내피 세포 특이적 Kif11 결손이 있는 마우스에서의 중증 망막 혈관 성장 지연[19].
게놈 편집 마우스 모델은 희귀 질환 기전 연구와 약물 유효성 평가에서 중요한 역할을 합니다. Cyagen은 희귀 질환에 대한 다양한 유전자 녹아웃(KO) 및 조건부(예: 조직 특이적) KO 모델 등 독자적으로 개발한 NDP, FZD4, LRP5 등의 수천 가지 게놈 편집 마우스 계통 품종(strain)을 제공하고 있습니다. 또한, 당사의 맞춤형 모델 개발 및 CRO 서비스는 니즈에 맞춰 조정할 수 있어, 이를 통해 연구 프로젝트를 신속하게 수행할 수 있습니다.
|
질환 |
표적 유전자 |
유형 |
|
가족성 삼출성 유리체 망막병증(Familial Exudative Vitreoretinopathy, FEVR) |
Ndp |
KO、CKO |
|
Fzd4 |
KO、CKO |
|
|
LRP5 |
KO、CKO |
|
|
Tspan12 |
KO、CKO |
|
|
Ctnna1 |
CKO |
|
|
Ctnnb1 |
KO、CKO |
|
|
Kif11 |
KO、CKO |
참고문헌:
[1]“Familial Exudative Vitreoretinopathy (FEVR).” EyeWiki. American Academy of Ophthalmology, 6 July 2023. https://eyewiki.aao.org/Familial_Exudative_Vitreoretinopathy_(FEVR).
[2]Ranchod TM, Ho LY, Drenser KA, Capone A Jr, Trese MT. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology. 2011 Oct;118(10):2070-5.
[3]Xiao H, Tong Y, Zhu Y, Peng M. Familial Exudative Vitreoretinopathy-Related Disease-Causing Genes and Norrin/β-Catenin Signal Pathway: Structure, Function, and Mutation Spectrums. J Ophthalmol. 2019 Nov 16;2019:5782536.
[4]Wang X, Chen J, Xiong H, Yu X. Genotype-phenotype associations in familial exudative vitreoretinopathy: A systematic review and meta-analysis on more than 3200 individuals. PLoS One. 2022 Jul 13;17(7):e0271326.
[5]Jia LY, Ma K. Novel Norrie disease gene mutations in Chinese patients with familial exudative vitreoretinopathy. BMC Ophthalmol. 2021 Feb 15;21(1):84.
[6]Luhmann UF, Lin J, Acar N, Lammel S, Feil S, Grimm C, Seeliger MW, Hammes HP, Berger W. Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest Ophthalmol Vis Sci. 2005 Sep;46(9):3372-82.
[7]Huang L, Lu J, Zhang L, Zhang Z, Sun L, Li S, Zhang T, Chen L, Cao L, Ding X. Whole-Gene Deletions of FZD4 Cause Familial Exudative Vitreoretinopathy. Genes (Basel). 2021 Jun 27;12(7):980.
[8]Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004 Mar 19;116(6):883-95.
[9]Lai MB, Zhang C, Shi J, Johnson V, Khandan L, McVey J, Klymkowsky MW, Chen Z, Junge HJ. TSPAN12 Is a Norrin Co-receptor that Amplifies Frizzled4 Ligand Selectivity and Signaling. Cell Rep. 2017 Jun 27;19(13):2809-2822.
[10]Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009 Oct 16;139(2):299-311
[11]Norwitz NG, Mota AS, Misra M, Ackerman KE. LRP5, Bone Density, and Mechanical Stress: A Case Report and Literature Review. Front Endocrinol (Lausanne). 2019 Mar 26;10:184.
[12]Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Jüppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML; Osteoporosis-Pseudoglioma Syndrome Collaborative Group. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001 Nov 16;107(4):513-23.
[13]Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Chan L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002 Apr 15;157(2):303-14.
[14]Kim SP, Frey JL, Li Z, Goh BC, Riddle RC. Lack of Lrp5 Signaling in Osteoblasts Sensitizes Male Mice to Diet-Induced Disturbances in Glucose Metabolism. Endocrinology. 2017 Nov 1;158(11):3805-3816.
[15]Li N, Xu Y, Li G, Yu T, Yao RE, Wang X, Wang J. Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: Case report and literature review. Medicine (Baltimore). 2017 May;96(20):e6914.
[16]Panagiotou ES, Sanjurjo Soriano C, Poulter JA, Lord EC, Dzulova D, Kondo H, Hiyoshi A, Chung BH, Chu YW, Lai CHY, Tafoya ME, Karjosukarso D, Collin RWJ, Topping J, Downey LM, Ali M, Inglehearn CF, Toomes C. Defects in the Cell Signaling Mediator β-Catenin Cause the Retinal Vascular Condition FEVR. Am J Hum Genet. 2017 Jun 1;100(6):960-968.
[17]He Y, Yang M, Zhao R, Peng L, Dai E, Huang L, Zhao P, Li S, Yang Z. Novel truncating variants in CTNNB1 cause familial exudative vitreoretinopathy. J Med Genet. 2023 Feb;60(2):174-182.
[18]L, Huang Y, Fei P, Yang Y, Zhang S, Xu H, Yuan Y, Zhang X, Zhu X, Ma S, Hao F, Sundaresan P, Zhu W, Yang Z. Catenin α 1 mutations cause familial exudative vitreoretinopathy by overactivating Norrin/β-catenin signaling. J Clin Invest. 2021 Mar 15;131(6):e139869.
[19]Wang Y, Smallwood PM, Williams J, Nathans J. A mouse model for kinesin family member 11 (Kif11)-associated familial exudative vitreoretinopathy. Hum Mol Genet. 2020 May 8;29(7):1121-1131.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



