

이런 환자는 만나본 적이 있나요? 정상적으로 성장하였던 영유아는 얼굴 이상, 성장 부진, 지적 발달 지연, 골격 기형, 신경계 이상, 심장 무리 등 증상을 보이게 됩니다. 이는 사실상 대사장애에 따른 복잡한 다계통 질환이며, 발병은 여러 유전자와 환경 요소간의 상호작용과 관련됩니다. 이런 질환은 보통 신경계, 내분비계, 소화계, 골격계 및 심혈관계 등 인체의 여러 계통에 영향을 미칩니다.
현재 이런 질환에 대한 치료법은 잠재적인 유전적 결함이 아닌 임상 증상에 중점을 두고 있기 때문에 환자는 평생 치료를 받아야 합니다. 대사 질환의 발병 매커니즘에 대한 이해 제고 및 잠재적인 치료법 모색을 위해 인간 질환을 전반적으로 복제할 수 있는 동물 모델을 구축할 필요가 있으며 이와 같은 대사 질환의 발병 및 잠재적인 치료 방법을 이해할 수 있는 중요한 도구를 제공해야 합니다.
MLⅡ
뮤코지질증Ⅱ


뮤코지질증 (Mucolipidosis)은 리소좀 효소 결핍으로 인해 뮤코다당류 분해장애가 발생하는 리소좀 희귀 질환입니다. 이는 세포 내, 혈액 및 결합 조직에 당 분자의 축적으로 이어져 여러 임상 표현형을 유발합니다. 이런 질환은Ⅰ-Ⅶ형 총 7가지 유형과 여러 하위 유형이 있습니다. 그중 뮤코지질증Ⅱ(mucolipidosis Ⅱ, MlⅡ)과 뮤코지방증 III alpha/beta(mucolipidosisⅢ α/β, MlⅢα/β)는 GNPTAB 유전자의 돌연변이에 의해 발생합니다. GNPTAB 유전자에 의해 암호화된 N-아세틸글루코사민-1-포스포트랜스퍼라제라는 리소좀 단백질의 글리코실화와 위치확인에 중요한 역할을 하기 때문에 GNPTAB 유전자의 돌연변이로 인한 효소 기능 손상은 리소좀의 정상적인 기능 수행에 영향을 끼칩니다. 증상으로는 성장 부진, 지적 발달 지연, 비정상적인 얼굴 특징, 골격 기형, 신경계 이상, 심장 무리로 인한 심부전 등이 있습니다[1].
Paton L 등이 구축한 Gnptab 결핍 마우스 모델은 인간 MLⅡ의 병리를 완전히 복제하여 성장 부진, 비정상적인 골격 및 얼굴 특징, 순환 리소좀의 효소 활성 증가, 세포 내 리소좀 저장 및 수명 단축 등 특징을 나타내고 있습니다. [2] Ko AR 등은 유전자 편집 기술로 모델 마우스의 Gnptab 유전자 제12~22번째 엑손을 제거하여 얻은 Gnptab-KO 마우스 역시 MLⅡ의 다양한 증상을 나타내 병리학적인 진전을 보였습니다. 또한 재조합 바이러스 벡터를 통한 모델 마우스에서의 Gnptab 유전자 발현은 뚜렷한 치료 효과를 얻을 수 있으며, 이는 해당 질환의 유전자 치료에 추가 참고 자료를 제공합니다[3].
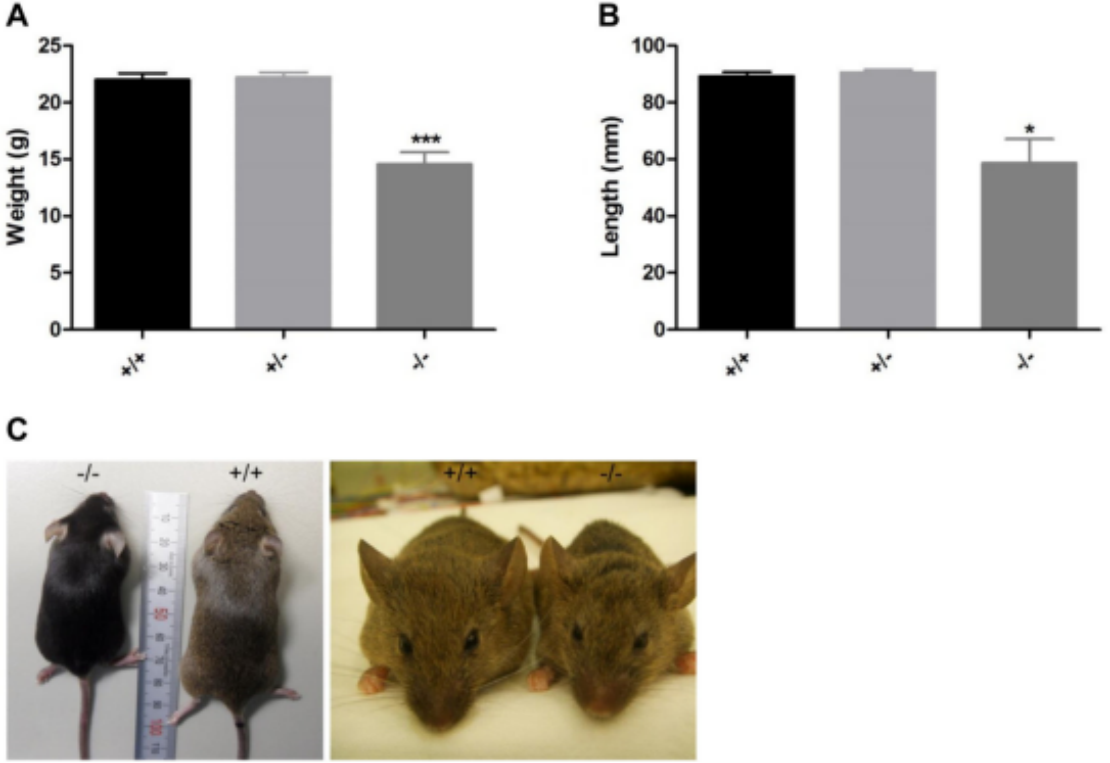
그림1 GNPTAB KO 마우스(-/-)에 성장 부진 및 얼굴 기형의 표현형 보임[3]
MDDGA3
근이영양증-영양실조형 당뇨병A3


근이영양증-영양실조형 당뇨병A3(MDDGA3)은 근육-눈-뇌 질환(MEB)이라고도 하고, 아미노산 대사효소의 유전자 돌연변이에 의해 발생한 희귀 선천성 근이영양증(CMD)입니다. 주요 특징은 출생 직후의 저긴장, 환자는 근이영양증, 중추신경계 이상 및 눈의 이상을 보입니다. MEB는 POMGNT1 유전자의 동형접합성 혹은 이형접합성 돌연변이에 기인되고 , POMGNT1 유전자에 의해 암호화된 0-만노스 β-1,2-N-아세틸글루코사민 전이효소(POMGnT1)가 0-만노스 글리코실 합성에 참여합니다. POMGnT1가 α-디스트로글리칸(α-DG)애 대한 산화, 글리코실화 변형은 근섬유막의 안정성, 액틴 세포골격과 세포외 기질의 연결에 매우 중요합니다. 따라서 POMGNT1 유전자의 돌연변이는 0-만노스 글리코실의 합성 장애를 일으켜, α-DG 글리코실의 감소로 이어져 MDDGA3를 유발할 수 있습니다[4]. 현재의 물리치료는 근력과 조정력 개선에, 항생제, 피질 호르몬, 비타민 B1, 비타민 B12 및 아데노신삼인산은 환자의 기능 회복에, 침구는 눈근육 마비 완화에 도움이 되는데 효과적인 완치법은 아직 없습니다.
모델 마우스에서 POMGnT1의 결핍은 근육, 눈 및 뇌의 발달 결함을 유발하며, 다른 증상으로는 선천성, 비진행성, 경증에서 중등도의 감각 신경성 난청과 달팽이관 말초 신경에 발생한 심각한 미엘린초 손상이 있습니다. 또한, POMGnT1 결핍 마우스의 망막에 뮬러세포와 성상세포 등 반응성 교세포 증식을 보였습니다[5-7]. 인간 MDDGA3 질환과 매우 유사한 증상발현을 나타낸 POMGnT1 결핍 마우스는 질환의 메커니즘 연구 및 약물 개발 등에 사용할 수 있습니다.
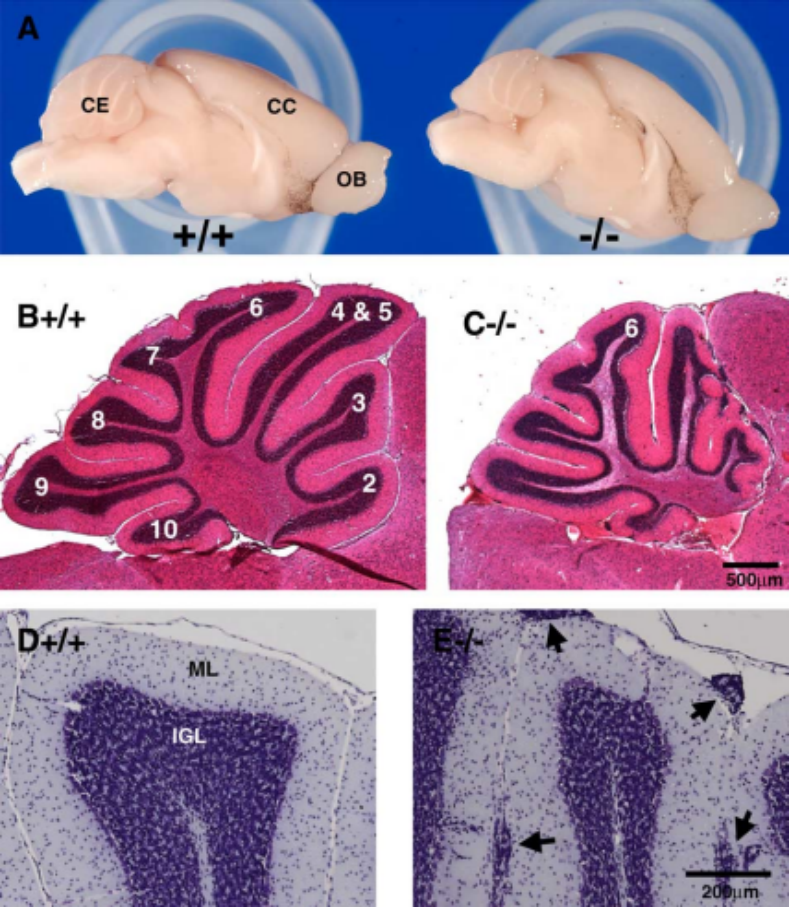
그림2 POMGnT1 결핍 마우스에 소뇌 위축 특징을 보임[5]
볼프람 증후군
DIDMOAD(요붕증, 당뇨병, 시신경 위축 및 난청)증후군


DIDMOAD(요붕증, 당뇨병, 시신경 위축 및 난청)증후군은 상염색체 열성 유전 질환으로 볼프람 증후군이리고도 합니다. 청소년 시기에 발생하는 당뇨병, 요붕증, 시신경 위축과 신경퇴화가 특징입니다. 국제질병분류초안(ICD-11)에서는 볼프람 증후군을 희귀 특정 당뇨병으로 분류하고 있습니다. 현재 볼프람 증후군의 진행을 늦추거나 되돌리 수 있는 효과적인 치료법은 없으므로, 임상 모니터링과 지지요법으로 환자의 고통을 완화하고 삶의 질을 개선하고 있습니다. 해당 질환은 주로 아동기와 성년기 초반에 발병하는 경향이 있고, 보통 15세 전에 당뇨병과 시력 문제가 나타나고, 뇌 기능 손상으로 단명할 수도 있습니다. 볼프람 증후군는 유전자의 돌연변이에 기인되고 협재 확인되는 관련 유전자는 2개가 있습니다. WFS1 유전자에 의해 발생되는 볼프람 증후군 1형과 WFS2 유전자에 의해 발생되는 볼프람 증후군 2형입니다[8].
Wfs1 녹아웃 마우스는 볼프람 증후군과 관련된 다양한 표현형 특성을 나타냅니다.당뇨병(고혈당, 내당능 장애, 췌도 기능 저하)[9-10], 시신경 위축(시신경 및 망막 신경절 세포의 퇴화로 인한 진행성 시력 상실)[11], 요붕증(항이뇨 호르몬 합성 또는 분비 결함으로 인한 다뇨 및 갈증), 신경계 이상(운동 협응 및 균형 장애, 행동 이상 및 정서 장애 동반)[12] 등이 있습니다. Wfs1 녹아웃 마우스에서 관찰된 이러한 표현형 특성은 볼프람 증후군 발병 매커니즘 이해 제고와 잠재적인 치료법 개발에 중요한 참고 자료를 제공합니다.
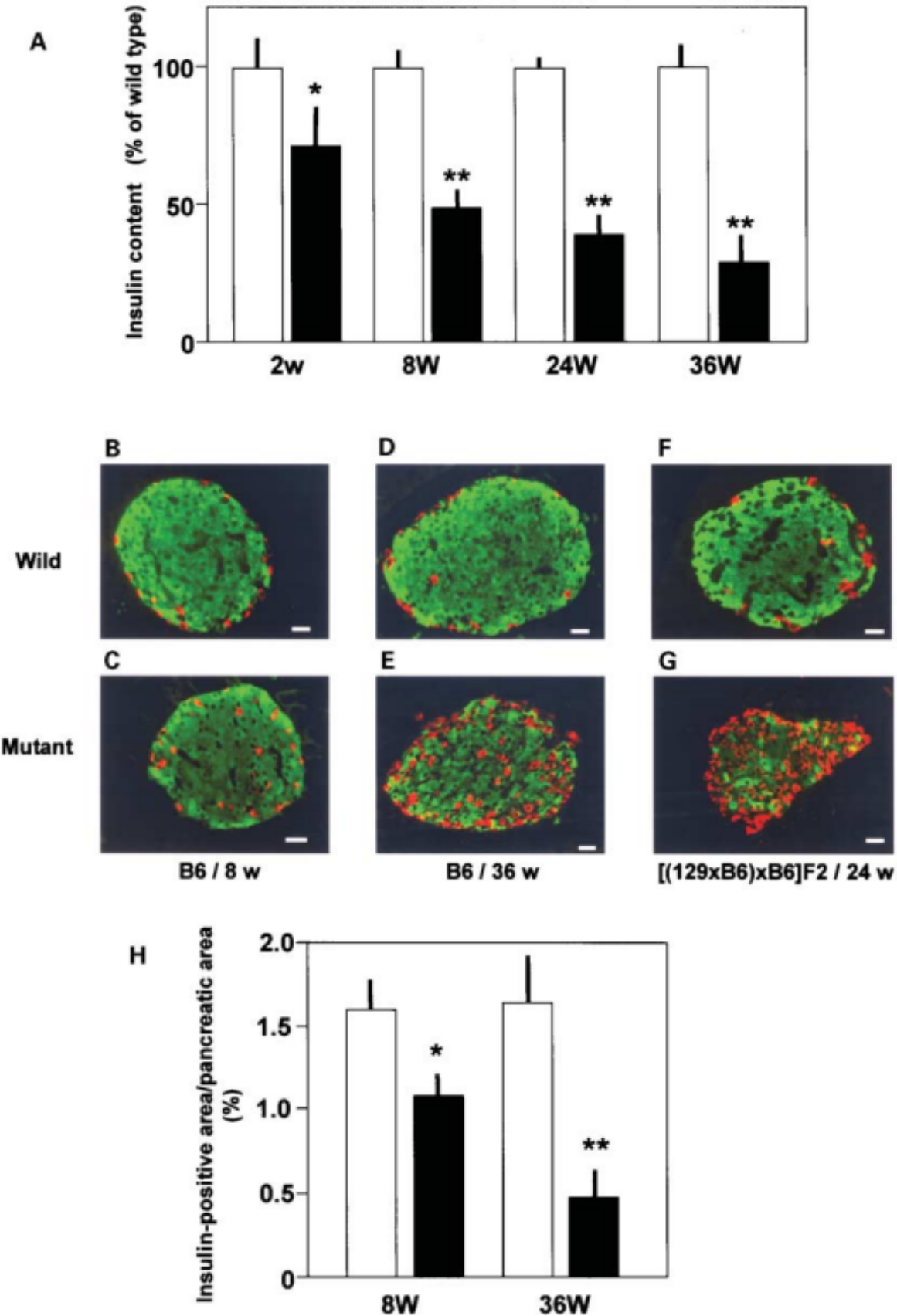
그림3 Wfs1 결핍 마우스에 진행성 췌장 β 세포 결함을 보임[9]
CLD
선천성 분비성 염화물 설사


선천성 분비성 염화물 설사(CLD)는 상염색체 열성으로 유전되는 희귀 질환으로 수양성 설사, 높은 대변의 염소 이온 농도, 대사성 알칼리증, 전해질 이상을 특징으로 보입니다. 현재의 치료법은 잠재적인 유전적 결함이 아닌 CLD 증상에 중점을 두고 있습니다. 조기 진단과 적극적인 대용소금 치료로 정상적인 성장 발달을 유지할 수 있고 좋은 예후를 얻을 수 있으며, 염화나트륨과 염화 칼륨을 사용하는 대용소금 치료 또한 환아에게 효과가 있습니다. 하지만 이같은 치료법음 잠재적인 유전적 결함을 근절할 수 없음으로 평생 치료를 받아야 합니다. CLD는 장세포의 막 단백질을 암호화하는 SLC26A3 유전자의 돌연변이에 의해 발생하며, 이는 장세포의 막 단백질의 기능적 결핍을 유발하여 위장관 내 물질들이 산성이 되고. 액체 흡수가 더 어려워지면서, 대량의 수양성 설사와 대사성 알칼리증으로 이어지고, 속발성 고혈압 신장질환 고알도스테론증을 동반합니다[13]. 일부 Slc26a3 KO 마우스는 출생 후 낮은 투과성으로 사망하게 되고, 살아 있는 마우스는 높은 농도의 염소를 포함하고 있는 설사, 성장 부진, 큰창자 확장, 결장 점막의 비정상적인 성장, 결장 움(crypt) 증식 영역의 큰 확장 및 혈장 내 알도르테론 증가 등 전형적인 CLD 임상 소견을 보였습니다.[14]
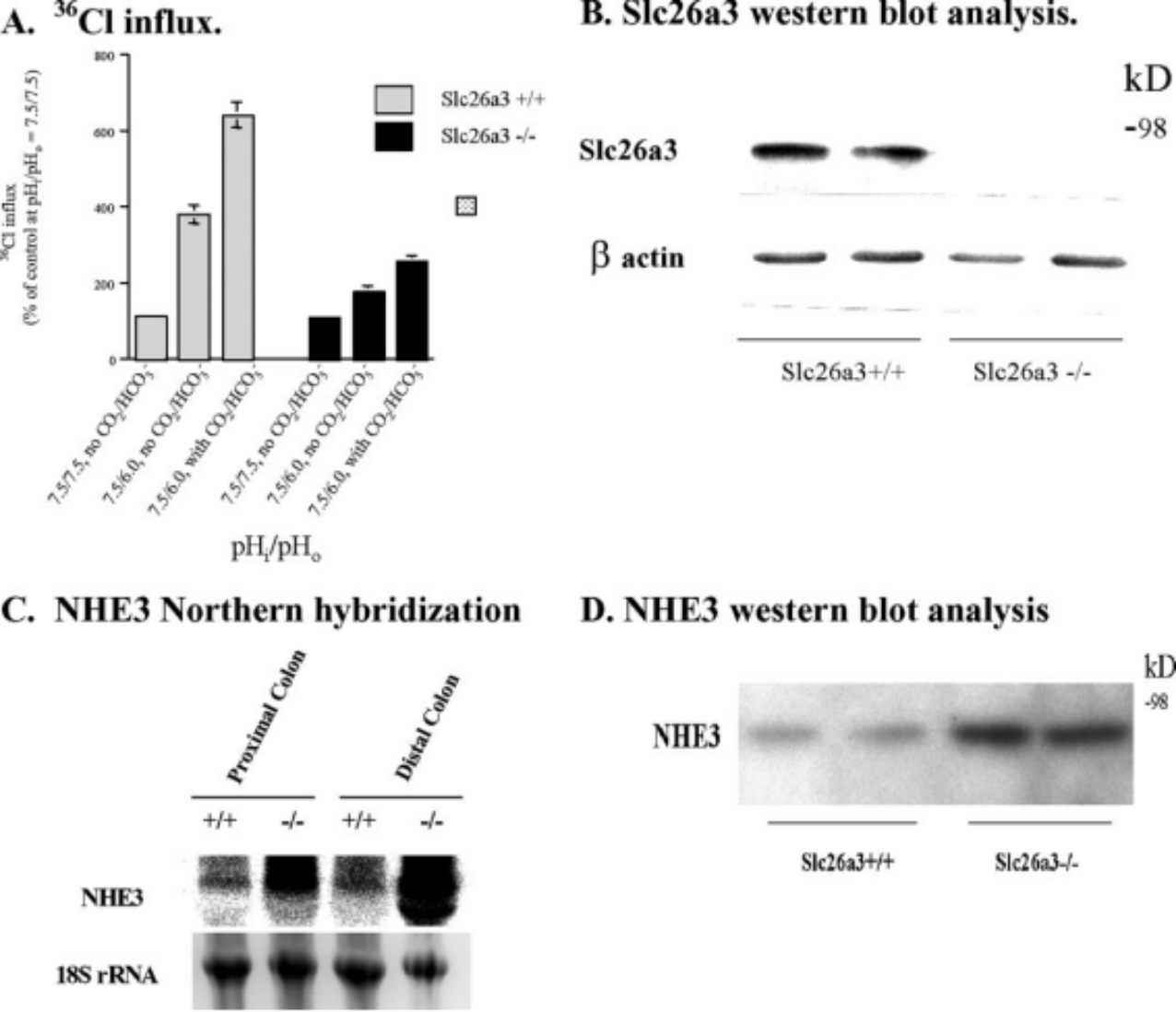
그림4 Slc26a3 KO 마우스 결장에서 이온 수송체의 발현 및 활성 결핍으로 인한 염화물 이온 조절 장애[14]
Cyagen 희귀 질환 연구 자원
유전자 편집 마우스
유전자 편집 마우스 모델은 희귀 질환 매커니즘 연구 및 약물 연구, 개발, 평가에 중요한 역할을 합니다. Cyagen는 수천개의 자체 개발한 유전자 편집 마우스 종류를 보유하고 있으며, GNPTAB, POMGNT1, WFS1, SLC26A3를 포함하여 희귀질환 연구에 사용하는 다양한 유전자 녹아웃 혹은 조건부 녹아웃 마우스 모델을 제공할 수 있습니다. 또한 과학연구 요구에 따라 전문화된 맞춤형 서비스를 제공하여 더 빠른 연구에 도움이 될 것입니다.
|
질환모델 |
발병 유전자 |
유형 |
|
뮤코지질증Ⅱ(MLⅡ) |
Gnptab |
KO, CKO |
|
근이영양증-영양실조형 당뇨병A3(MDDGA3) |
Pomgnt1 |
KO, CKO |
|
DIDMOAD(요붕증, 당뇨병, 시신경 위축 및 난청)증후군 |
Wfs1 |
KO, CKO |
|
선천성 분비성 염화물 설사(CLD) |
Slc26a3 |
KO, CKO |
Cyagen맞춤형 모델 서비스
대사 모델 추천
인구 고령화와 비만의 증가에 따라서 대사성 질환의 발병률은 해마다 증가세를 보이고 있고, 인간의 건강을 심각하게 해치고 있습니다. Cyagen약물 스크리닝 및 평가 마우스 모델 플랫폼은 Ldlr KO (em), Lep KO, Uox-KO (Prolonged), Atp7b KO, Foxj1 KO 등 다양한 유형의대사 관련 유전자 편집 모델을 제공하여 신약 개발에 도움을 줍니다.
대사 및 심혈관 질환 특선 모델-유전자 편집
|
제품코드 |
제품명 |
품계 배경 |
품계 배경 |
|
COO1067 |
APOE |
C57BL/6N |
죽상동맥경화증 |
|
C001291 |
B6-db/db |
C57BL/6J |
고혈당 및 비만 |
|
C001392 |
Ldlr-KO(em) |
C57BL/6J |
가족성 고콜레스테롤혈증 |
|
COO1368 |
B6-ob/ob(Lep KO) |
C57BL/6J |
II형 당뇨병 및 비만 |
|
C001232 |
Uox KO |
C57BL/6J |
고요산혈증 |
|
C001393 |
Uox-KO(Prolonged) |
C57BL/6J |
고요산혈증 |
|
COO1267 |
Atp7b KO |
C57BL/6N |
구리대사장애질환(윌슨병) |
|
C001265 |
Foxj1 KO |
C57BL/6N |
원발성 섬모 이상운동증 |
|
C001266 |
Usp26 KO |
C57BL/6N |
클라인펠터 증후군 |
|
C001273 |
Fah KO |
C57BL/6N |
타이로신혈증 I형 |
|
C001383 |
Alb-Cre/LSL-hLPA |
C57BL/6N |
심혈관 목표 환부 |
|
C001421 |
B6-hGLP-1R |
C57BL/6N |
대사 목표 환부 |
|
C001400 |
B6J-hANGPTL3 |
C57BL/6J |
대사 목표 환부 |
대사 및 심혈관 질환 특선 모델-유도 모델
|
폐고혈압 모델 |
심혈관질환 모델 |
동맥경화 모델 |
말초혈관질환 모델 |
|
알코올성 지방간염 모델 |
비알코올성 지방간염 모델 |
CCL4에 의한 급성 간손상 모델 |
만성 간손상 모델 |
|
당뇨병 및 합병증 모델 |
비만 모델 |
신장질환 모델 |
뇌졸중 모델 |
참고 문헌:
[1]Leroy JG, Cathey SS, Friez MJ. GNPTAB-Related Disorders. 2008 Aug 26 [updated 2019 Aug 29]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023.
[2]Paton L, Bitoun E, Kenyon J, Priestman DA, Oliver PL, Edwards B, Platt FM, Davies KE. A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J Biol Chem. 2014 Sep 26;289(39):26709-26721. doi: 10.1074/jbc.M114.586156. Epub 2014 Aug 8.
[3]Ko AR, Jin DK, Cho SY, Park SW, Przybylska M, Yew NS, Cheng SH, Kim JS, Kwak MJ, Kim SJ, Sohn YB. AAV8-mediated expression of N-acetylglucosamine-1-phosphate transferase attenuates bone loss in a mouse model of mucolipidosis II. Mol Genet Metab. 2016 Apr;117(4):447-55.
[4]Shenoy AM, Markowitz JA, Bonnemann CG, Krishnamoorthy K, Bossler AD, Tseng BS. Muscle-Eye-Brain disease. J Clin Neuromuscul Dis. 2010 Mar;11(3):124-6.
[5]Liu J, Ball SL, Yang Y, Mei P, Zhang L, Shi H, Kaminski HJ, Lemmon VP, Hu H. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech Dev. 2006 Mar;123(3):228-40.
[6]Morioka S, Sakaguchi H, Mohri H, Taniguchi-Ikeda M, Kanagawa M, Suzuki T, Miyagoe-Suzuki Y, Toda T, Saito N, Ueyama T. Congenital hearing impairment associated with peripheral cochlear nerve dysmyelination in glycosylation-deficient muscular dystrophy. PLoS Genet. 2020 May 26;16(5):e1008826.
[7]Takahashi H, Kanesaki H, Igarashi T, Kameya S, Yamaki K, Mizota A, Kudo A, Miyagoe-Suzuki Y, Takeda S, Takahashi H. Reactive gliosis of astrocytes and Müller glial cells in retina of POMGnT1-deficient mice. Mol Cell Neurosci. 2011 Jun;47(2):119-30.
[8]Urano F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr Diab Rep. 2016 Jan;16(1):6.
[9]Ishihara H, Takeda S, Tamura A, Takahashi R, Yamaguchi S, Takei D, Yamada T, Inoue H, Soga H, Katagiri H, Tanizawa Y, Oka Y. Disruption of the WFS1 gene in mice causes progressive beta-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum Mol Genet. 2004 Jun 1;13(11):1159-70.
[10] Ivask M, Volke V, Raasmaja A, Kõks S. High-fat diet associated sensitization to metabolic stress in Wfs1 heterozygous mice. Mol Genet Metab. 2021 Sep-Oct;134(1-2):203-211.
[11] Waszczykowska A, Zmysłowska A, Braun M, Ivask M, Koks S, Jurowski P, Młynarski W. Multiple Retinal Anomalies in Wfs1-Deficient Mice. Diagnostics (Basel). 2020 Aug 19;10(9):607.
[12] Visnapuu T, Raud S, Loomets M, Reimets R, Sütt S, Luuk H, Plaas M, Kõks S, Volke V, Alttoa A, Harro J, Vasar E. Wfs1-deficient mice display altered function of serotonergic system and increased behavioral response to antidepressants. Front Neurosci. 2013 Jul 31;7:132.
[13] Di Meglio L, Castaldo G, Mosca C, Paonessa A, Gelzo M, Esposito MV, Berni Canani R. Congenital chloride diarrhea clinical features and management: a systematic review. Pediatr Res. 2021 Jul;90(1):23-29.
[14] Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem. 2006 Dec 8;281(49):37962-71.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



