

척수성 근위축증(SMA)은 척수의 운동신경세포가 상실되는 유전질환으로 중추신경계, 말초신경계, 자율근육운동 등에 영향을 미칩니다.
이 글에서 SMN1의 기능을 검토하고 척추성근육위축증에 대한 표적 유전자 치료에서 SMN1의 역할을 탐구하여 SMN1 유전자 연구에 대해 알아보겠습니다.
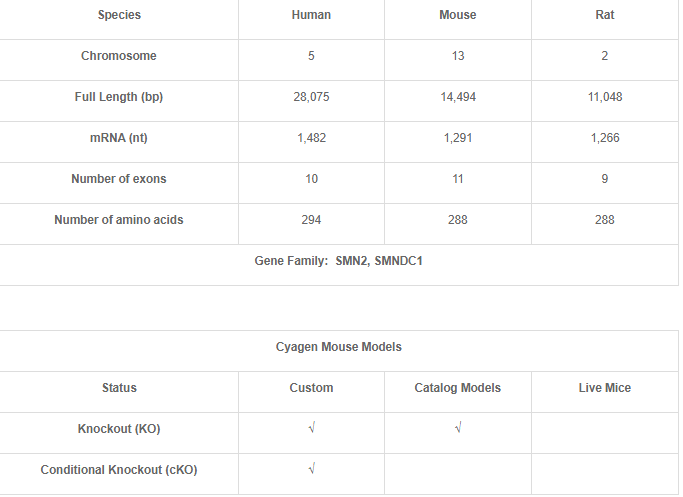
*: √로 체크된 것은 Cyagen AI Knock-Out Mouse Model eBank에서 이용할 수 있는 해당 모델을 나타낸다.
Survival motor neuron gene 1(SMN1)은 동일한 이름의 단백질을 암호화합니다. 이 단백질은 인간 유전 질환인 척수성 근위축증(Spinal Muscular Atrophy, SMA)과 밀접한 관련이 있으며, 대부분의 경우 2세 이전에 신생아 사망을 유발합니다. SMN1의 single copy 비활성화(무증상) 현상은 아시아인 인구에서 약 1/50이며, 이는 1/10000 정도(변이 빈도는 인종 및 지역에 따라 다름)의 신생아 이환율을 초래합니다. SMN1은 spliceosome의 구성 요소이며, spliceosome complex는 작은 핵 리보핵단백(small nuclear ribonucleoprotein, snRNPs)의 조립에 촉매 역할을 하므로 pre-mRNA의 splicing에 중요한 역할을 합니다. 이름에서 알 수 있듯이 이 단백질은 운동 뉴런의 생존을 유지하는 데 필수적입니다.
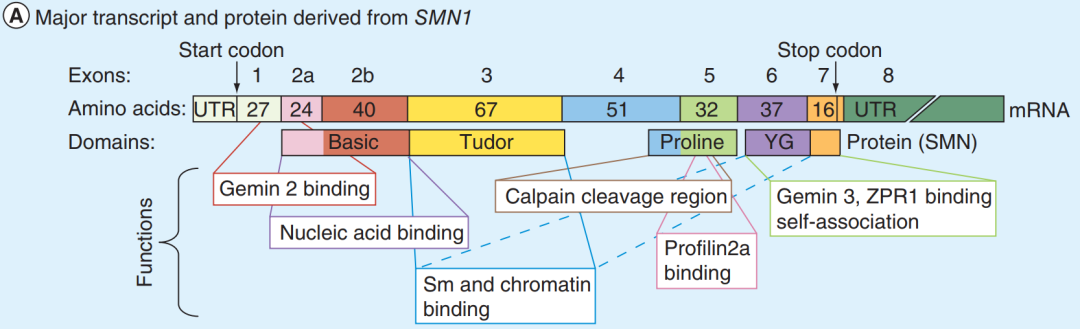
Figure 1: SMN1 Structure and Function. The figure shows the linearized structural regions of mature mRNA transcript and protein from human SMN1. Arrows indicate the locations of the start and stop codons. Exon regions are separated into boxes, with the number of amino acids noted in each. Different functional regions of the gene are annotated.1
인간 SMN 유전자는 SMN1과 SMN2로 구분됩니다. SMN1 유전자는 텔로미어 쪽에 위치하며 전사 후 전장 mRNA를 생성합니다. SMN2 유전자는 centromere 쪽에 위치합니다. SMN2 유전자와 SMN1 유전자는 엑손 스플라이싱 인핸서(Exonic splicing enhancer, ESE)에 하나의 뉴클레오티드 차이가 있어 전사된 SMN2가 일곱 번째 엑손을 잃고 잘린 SMN 단백질을 암호화합니다. 잘린 SMN 단백질은 기능이 없으며 세포에서 빠르게 분해됩니다. 생리학적 조건에서 SMN2 mRNA의 일곱 번째 엑손은 경우에 따라 완전히 결실되지 않는 경우도 있습니다. SMN2 유전자는 여전히 정상적인 기능을 가진 SMN을 암호화할 수 있는 일부(10% ~ 15%)의 전장 mRNA를 생성할 수 있습니다. 그러나 일부 SMN2의 전사(약 10 ~ 15 %)는 봉쇄를 뚫고 완전한 mRNA를 생성해 기능성 단백질을 합성할 수 있습니다. 연구에 따르면 SMA의 95%는 SMN1 유전자의 돌연변이로 인해 발생하며 SMA 환자에서 SMN1 유전자의 결실로 충분한 SMN 단백질이 생성될 수 없습니다. 질병 상태에서 체내 SMN 단백질은 주로 SMN2 유전자로부터 유래되며, SMN2 유전자의 일부만 기능성 SMN 단백질을 생산할 수 있기 때문에 SMA는 주로 체내 SMN 단백질이 부족하여 발생합니다.
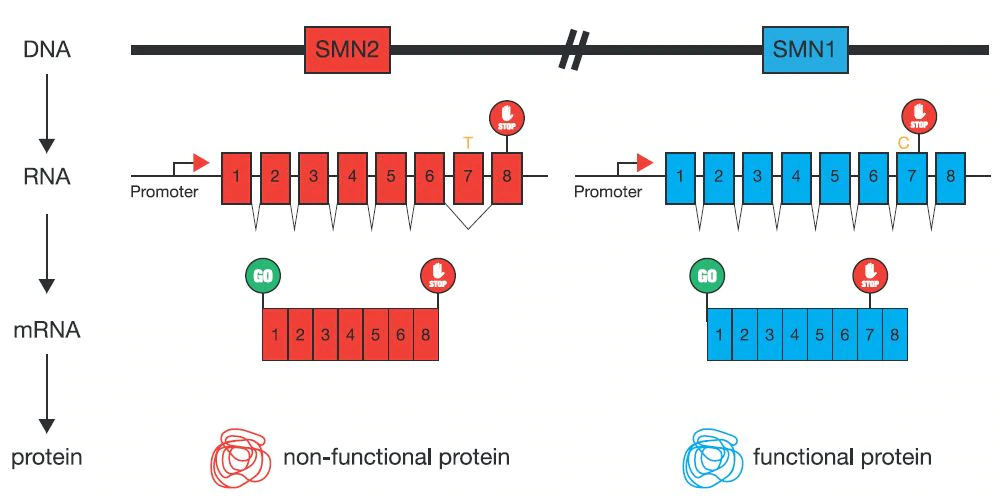
Figure 2: SMN1 and SMN22
앞서 언급한 SMA의 발병 메커니즘에 따르면 SMA의 치료 방안은 두 가지가 있습니다. 첫 번째 전략은 벡터를 통해 SMN1 정상 인코딩 유전자를 직접 도입하는 것인데, 이는 FDA가 승인한 약물인 Zolgensma(그림 3의 왼쪽에 표시된 전략)에 의해 채택된 치료법입니다. 이 방법은 또한 세 번째로 출시된 AAV 매개 유전자 치료 방안이 되었습니다. 두 번째 전략은 안티센스 뉴클레오타이드를 통해 환자의 신체 내에 여전히 존재하는 SMN2의 정상적인 발현을 촉진하는 것입니다(엑손 7의 스 플라이싱 억제). 가까운 장래에 유전자 치료가 SMA 환자에게 좋은 소식을 전하고 질병을 완전히 치료할 수 있기를 바랍니다.
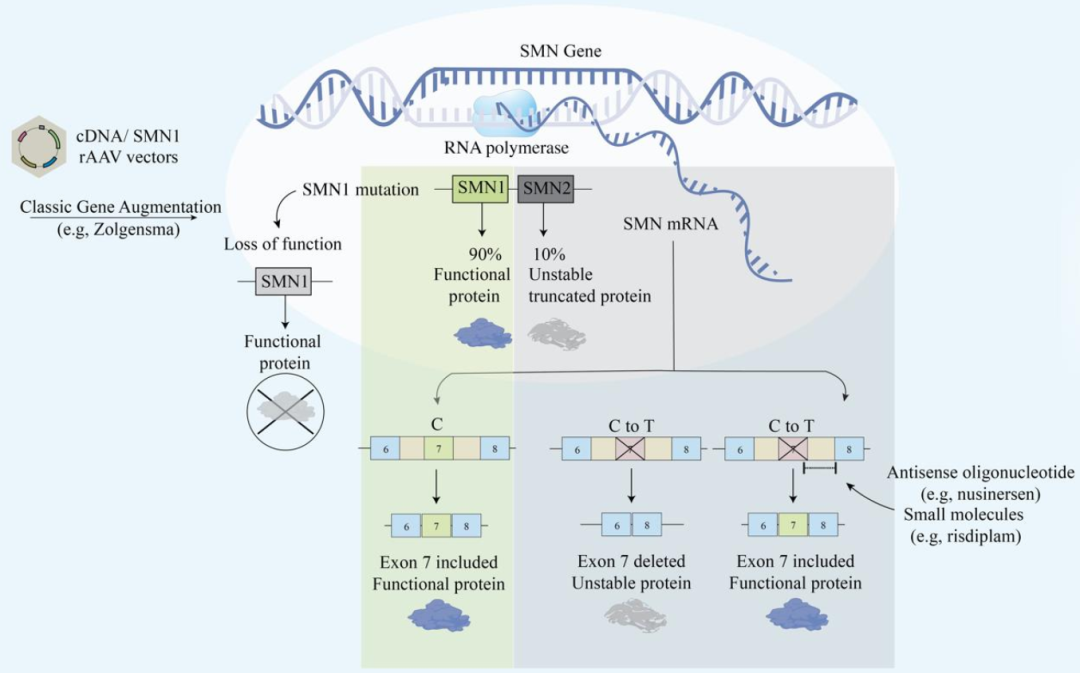
Figure 3: SMA Gene Therapy3
지난 수십 년 동안 유전자 치료는 유전성 질환 치료에서 상당한 진전을 이루었습니다. 유전체학 또는 단백질 체학으로 질병을 유발하는 유전자를 찾아내고, 세포 수준 또는 동물 수준 검증과 결합하는 패턴으로 발표되는 좋은 논문들이 많이 있습니다.
지난 10 년 동안 Cyagen은 유전자 치료 요법을 검증하기 위해 수백 개의 연구팀에 많은 유전자 변형 동물 모델 및 바이러스 패키징 서비스를 제공했습니다. 2020년 말까지, Nature, Cell, Science 등 유명한 저널을 비롯하여 Cyagen의 제품 및 서비스를 인용한 논문은 총 4,750건이고 IF(impact factor) 합계는 20,854이며 인용 횟수는 25,043 회가 누적되었습니다. Cyagen은 유전자 치료의 임상전환에 도움을 주는 것을 목표로 하고 있으며, 유전자 치료 연구 분야에 깊이 연구해 왔습니다. 또한 Cyagen은 실험 검증을 위한 유전자 변형 동물 모델과 바이러스 패키징 서비스를 제공할 수 있어 연구자들이 점수가 높은 논문을 발표할 수 있도록 도와주고, 유전자 치료 연구 및 임상전환에 도움을 줍니다. 관심이 있으시거나 문의 사항이 있으시면 +86 20-31601779 또는 service-apac@cyagen.com으로 연락 부탁드립니다.
References:



영업일 기준 1-2일 내에 답변해 드리겠습니다.



