

인간 최대 유전자의 연구를 위한 마우스 모델
듀센 근이영양증(DMD)을 앓고 있는 아이들이 달리고, 놀고, 퇴행성 근육 질환에 대한 두려움 없이 살아갈 수 있는 세상을 상상해 보세요. 이러한 미래는 더 이상 꿈이 아닙니다. 유전자 치료의 획기적인 발전과 인간화 마우스 모델의 등장으로, 듀센 근이영양증(DMD) 치료 분야는 전례 없는 변화를 맞이하고 있습니다. 본문에서 이 작은 동물 모델들이 어떻게 치료법 개발을 가속화하고 있는지 함께 살펴보겠습니다.
유전질환인 듀센 근이영양증(DMD) 연구의 최전선: 유전자 변형 동물 모델이 질환 해석과 치료 개발에서 수행하는 중요한 역할을 탐구합니다.
듀센 근이영양증(DMD) 이해하기: 치명적인 희귀 유전 질환
듀센 근이영양증(DMD)은 주로 남아에게 영향을 미치는 심각한 유전성 근육 질환으로, 발생률은 5,000명 중 1명입니다.[1] 이 질환은 DMD 유전자 돌연변이로 인해 발생합니다. DMD 유전자는 근육의 구조와 안정성에 필수적인 단백질인 디스트로핀(Dystrophin)을 인코딩합니다. 디스트로핀(Dystrophin)의 결핍은 점진적인 근육 퇴화와 근력 약화를 초래하며, 결국 평생 휠체어를 의존해야 하고 호흡 부전으로 이어질 수 있습니다.
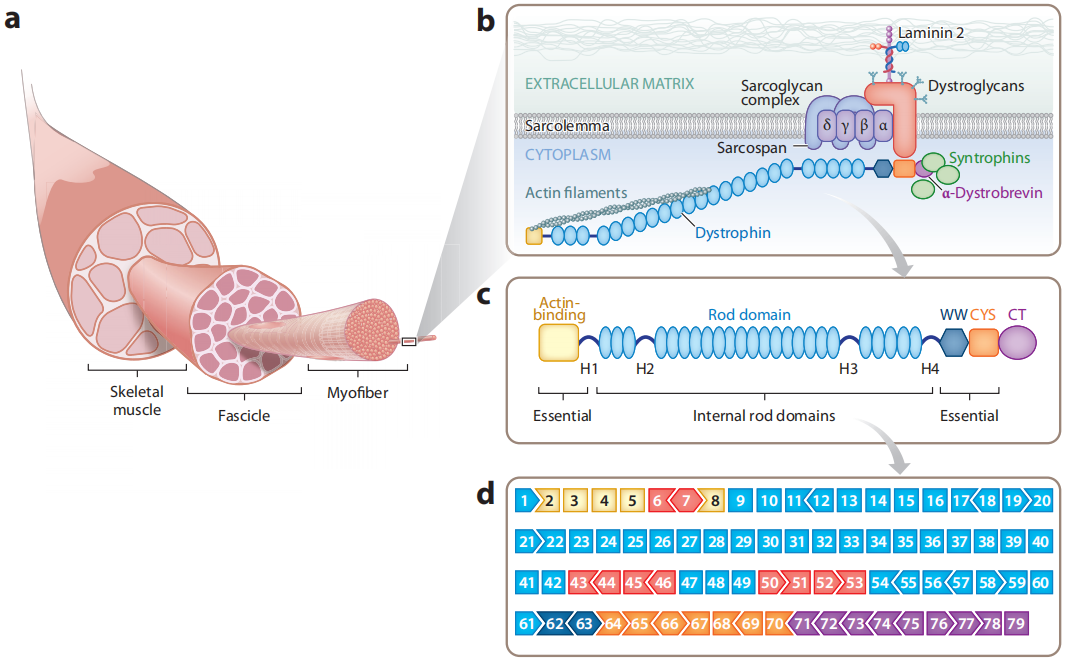
그림 1. 디스트로핀(Dystrophin): 근육 구조를 안정시키고 정상적인 근육 수축을 보장합니다. [1]
DMD 유전자 치료제 개발에서의 크기 장벽을 극복합니다
DMD 유전자는 인간 몸에서 알려진 가장 큰 단백질 코딩 유전자이며, 길이가 약 2.4Mb입니다. 75% 이상의 환자들이 DMD 유전자에서 대규모 결실이나 무의미 돌연변이를 가지고 있으며, 이는 주로 엑손 3-9와 엑손 45-55에 집중되어 있습니다. 이러한 돌연변이는 디스트로핀(Dystrophin) 결핍을 초래하고, 디스트로핀-당단백질 복합체(DGC)의 분해를 유발하여, 액틴과 세포외 기질 간의 상호작용을 방해하고, 근섬유 간의 결합을 약화시켜 근육 손상에 취약하게 만듭니다. 이 근육의 취약성은 결국 근육의 진행성 퇴화, 이동 능력 상실과 심근병증의 발병으로 이어집니다.[2]
기존의 아데노연관바이러스(AAV) 매개 유전자 치료법은 가능성을 보였지만, 제한된 벡터 용량으로 인해 전장 DMD 유전자를 전달할 수 없는 한계를 가지고 있습니다. 따라서 현재의 AAV 치료법은 훨씬 더 작은 버전의 디스트로핀(Dystrophin)은 전달하는 방법에 의존하고 있습니다. 이 방법이 바로 마이크로 디스트로핀(Micro-Dystrophin) “미니 버전” 유전자 치료 접근법입니다.[1-3]
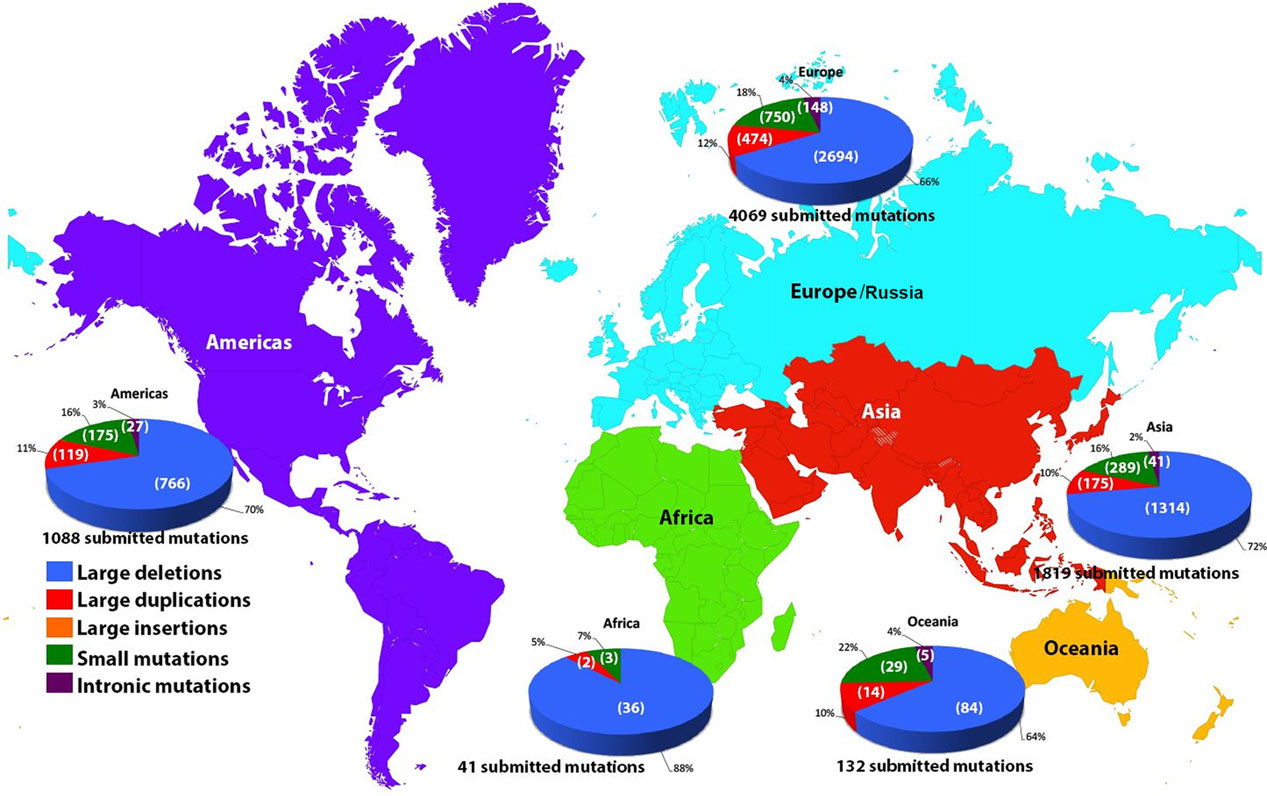
그림 2. DMD 돌연변이의 다양한 유형과 복잡성: 대규모 결실이 가장 흔히 발생하여 유전자 치료법 개발에 어려움을 초래합니다. [4]
마이크로 디스트로핀(Micro-Dystrophin) 유전자 치료의 한계: 면역원성의 위험
마이크로 디스트로핀(Micro-Dystrophin) 기반 치료는 여러 장점을 가지고 있지만, 여전히 면역원성(immunogenicity)과 관련된 잠재적 위험이 존재합니다. 일부 듀센 근이영양증(DMD) 환자는 유전자 결실로 인해 디스트로핀(Dystrophin)의 특정 항원결정기(epitope)를 잃어버린 상태입니다. 만약 유전자 치료법에 사용되는 마이크로 디스트로핀(Micro-Dystrophin)이 이 “결실된 영역”을 포함한다면, 환자의 면역계는 이를 비자가 항원(Non-self epitope)으로 인식하여 면역 거부 반응을 유발하고, 염증성 조직 손상을 초래할 수 있습니다.[5,6] 또한, 이러한 아데노연관바이러스(AAV) 기반 치료법은 일반적으로 높은 바이러스 용량이 필요하며, 이는 부작용 및 조직 독성의 위험을 높이는 요인이 됩니다.
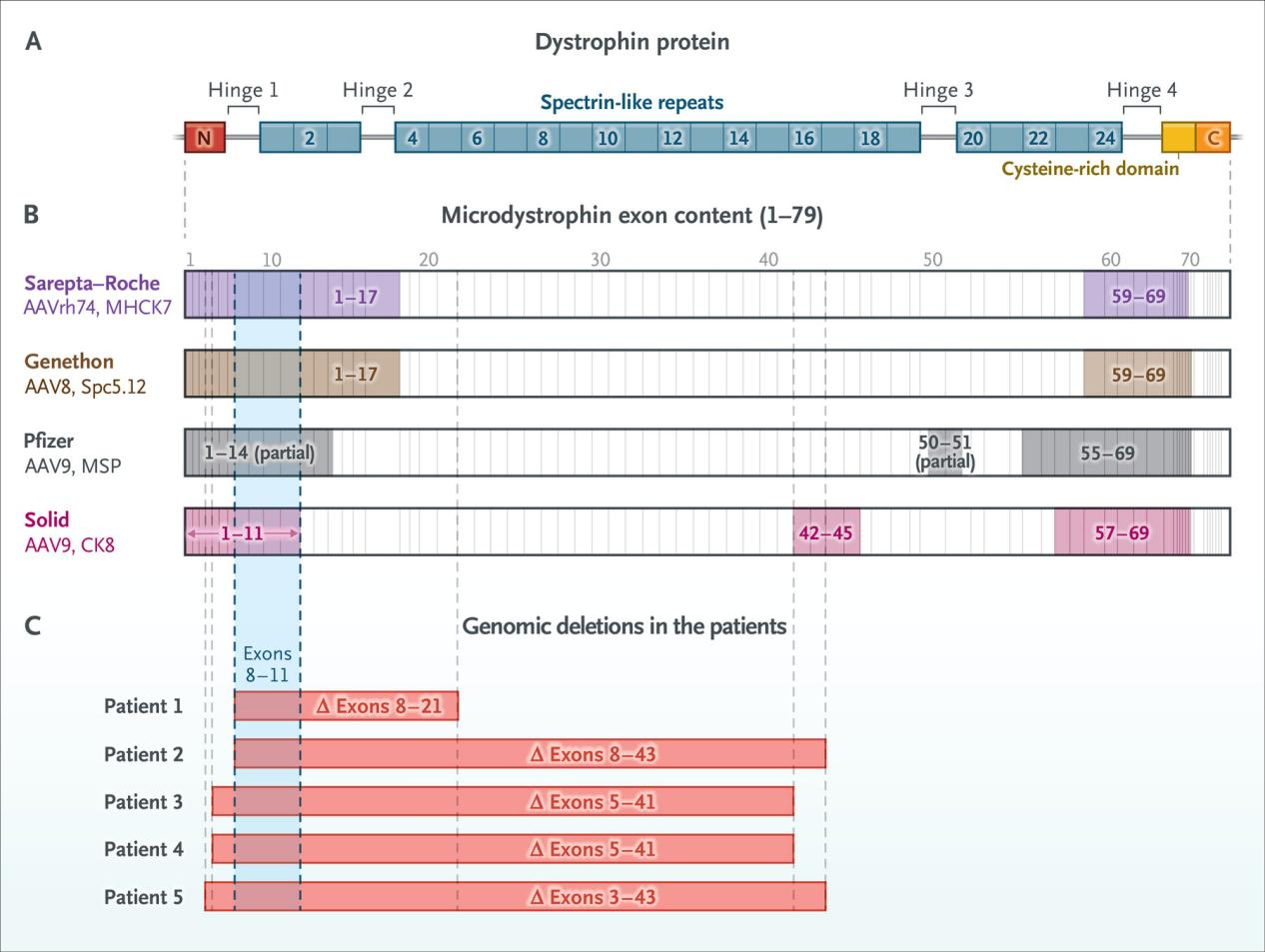
그림 3. AAV 유전자 치료는 면역원성 문제에 직면해 있으며, 일부 환자는 심각한 면역 이상 반응을 발생합니다. [6]
Exon Skipping 치료법: 듀센 근이영양증(DMD)을 위한 새로운 접근법
Exon Skipping 치료법은 유망한 대안으로 부상하고 있습니다. 이 치료법은 안티센스 올리고뉴클레오타이드(AON, antisense oligonucleotide)을 사용하여 pre-mRNA splicing 과정을 조절함으로써, 변이되거나 결실된 DMD 유전자의 엑손(exon)을 우회하도록 유도합니다. 이를 통해 mRNA의 리딩 프레임(reading frame)이 회복되며, 결과적으로 기능이 유지되는 짧은 형태의 디스트로핀(Dystrophin) 단백질이 생성될 수 있습니다.
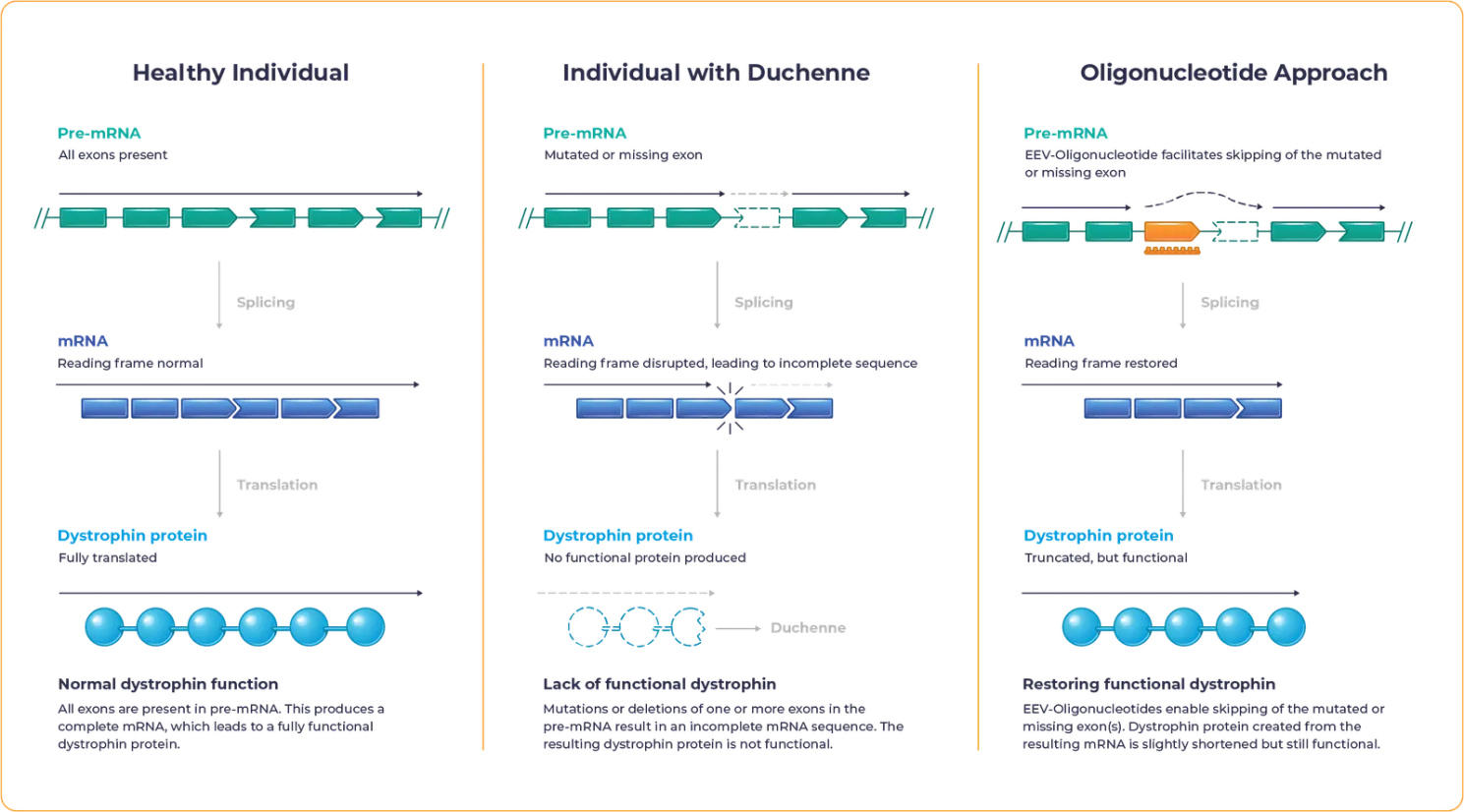
그림 4. 듀센 근이영양증(DMD) 치료를 위한 Exon Skipping 치료 기전 [7]
현재 엑손(exon) 45, 51, 53을 타겟으로 한 4종의 안티센스 올리고뉴클레오타이드(AON) 치료제가 FDA 승인을 받아 일부 환자들에게 새로운 희망을 제공하고 있습니다.[8] 그러나 여전히 많은 환자는 적응증에 포함되지 않아 치료 혜택을 받지 못하고 있습니다. 최근 연구에 따르면, 단일 또는 다중 Exon Skipping 전략을 통해 더 넓은 환자군에게 치료 혜택을 제공할 수 있습니다.
예를 들어, 엑손(exon) 51을 타겟으로 하는 Exon Skipping 전략은 유전자 대규모 결실(large deletion) 환자의 17.2%에 효과가 있으며, 엑손(exon) 45~55 또는 3~9를 타겟으로 하는 다중 Exon Skipping 전략은 각각 70.6%, 19.2%의 유전자 대규모 결실(large deletion) 환자에게 적용하며, 치료 대상 확대에 기여할 수 있습니다.[8]
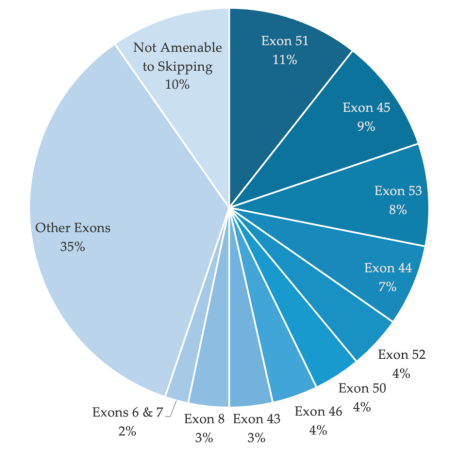
그림 5. Exon Skipping 전략은 다양한 DMD 유전자 돌연변이를 폭넓게 포괄할 수 있는 적용 가능성을 보여줍니다. [8]
Exon Skipping 치료법을 이론에서 임상 적용으로 성공적으로 전환하려면 안전성 및 효능 평가를 위한 유용한 전임상 동물 모델이 필요합니다. Exon Skipping 치료법은 인간 DMD 유전자를 타겟으로 하기 때문에 인간 DMD 유전자를 발현하는 인간화 DMD 마우스 모델은 안티센스 올리고뉴클레오타이드(AON) 후보물질의 전임상 시험에 이상적인 연구 모델입니다.
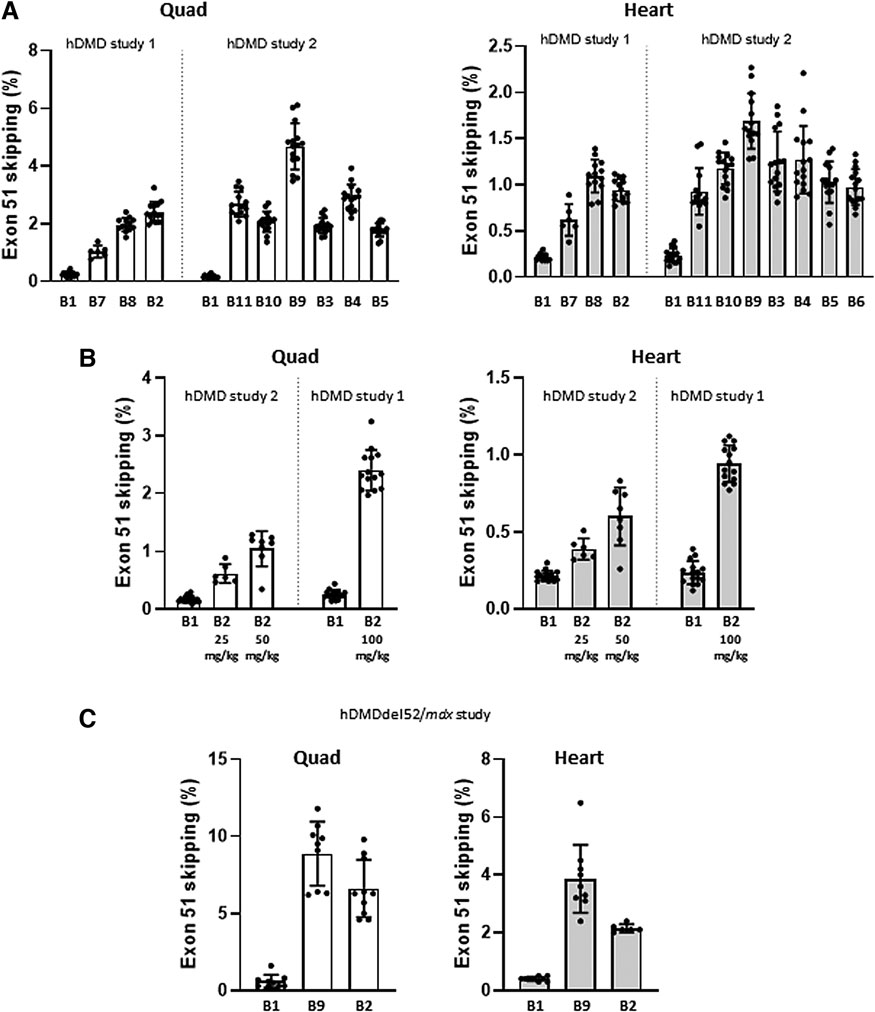
그림 6. 안티센스 올리고뉴클레오타이드(AON) 후보 물질의 In Vivo 스크리닝을 위한 인간화 DMD 마우스 모델. [9]
Cyagen의 인간화 DMD 마우스 모델로 치료제 개발을 앞당기세요
듀센 근이영양증(DMD) 치료제 연구를 가속하기 위해, Cyagen은 HUGO-GT™ (Humanized Genomic Ortholog for Gene Therapy) 플랫폼을 통해 여러 가지 인간화 DMD 마우스 모델을 개발하였습니다. 이 모델들은 인간 DMD 유전자에서 빈번하게 발견되는 변이 영역을 포함하고 있어, 연구자들이 Exon Skipping 치료제 개발을 빠르고 효율적으로 진행할 수 있는 유용한 전임상 연구 모델입니다.
|
제품 번호 |
품종 계통 |
인간화/돌연변이 전략 |
|
I001224 |
B6-hDMD(E8-30) |
Humanized Exons 8-30 |
|
I001204 |
B6-hDMD(E44-45) |
Humanized Exons 44-45 |
|
I001133 |
B6-hDMD(E49-53) |
Humanized Exons 49-53 |
|
I001208 |
B6-hDMD(E44-45)*Del E44 |
Humanized Exons 44-45 with Exon 44 Deletion |
|
TBD |
B6-hDMD(E44-45)*c.6438+2 T to A |
Humanized Exons 44-45 with c.6438+2 T to A Mutation |
|
TBD |
B6-hDMD(E49-53)*Del E50 |
Humanized Exons 49-53 with Exon 50 Deletion |
|
C001518 |
DMD-Q995*(C57BL/6) |
Mdx Point Mutation (p.Q995X, c.C2983T), C57BL/6 Background |
|
TBD |
DMD-Q995*(DBA/2) |
Mdx Point Mutation (p.Q995X, c.C2983T), DBA/2 Background |
일부 인간화 마우스 모델에 대한 검증 데이터는 다음과 같습니다.
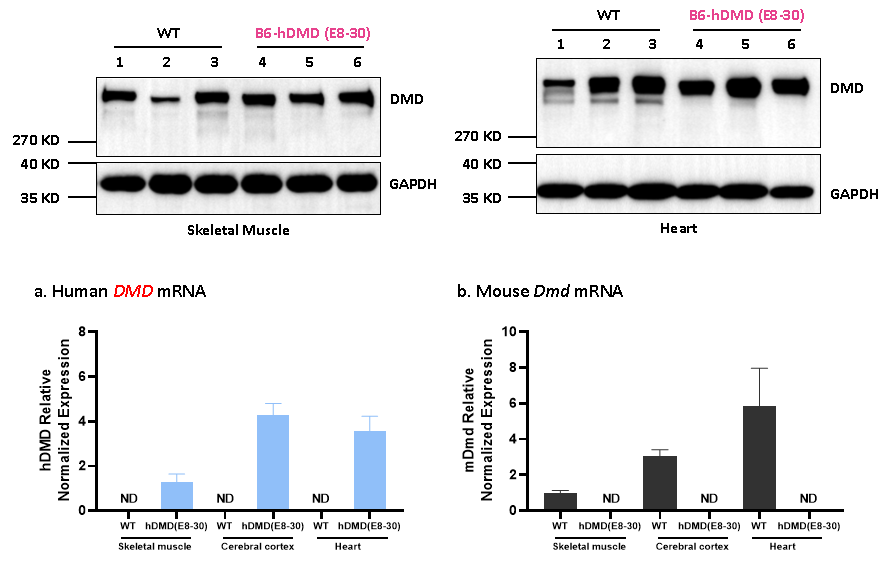
그림 7. 인간 DMD 유전자와 전장 디스트로핀(Dystrophin) 단백질의 발현에 성공한 B6-hDMD(E8-30) 마우스.
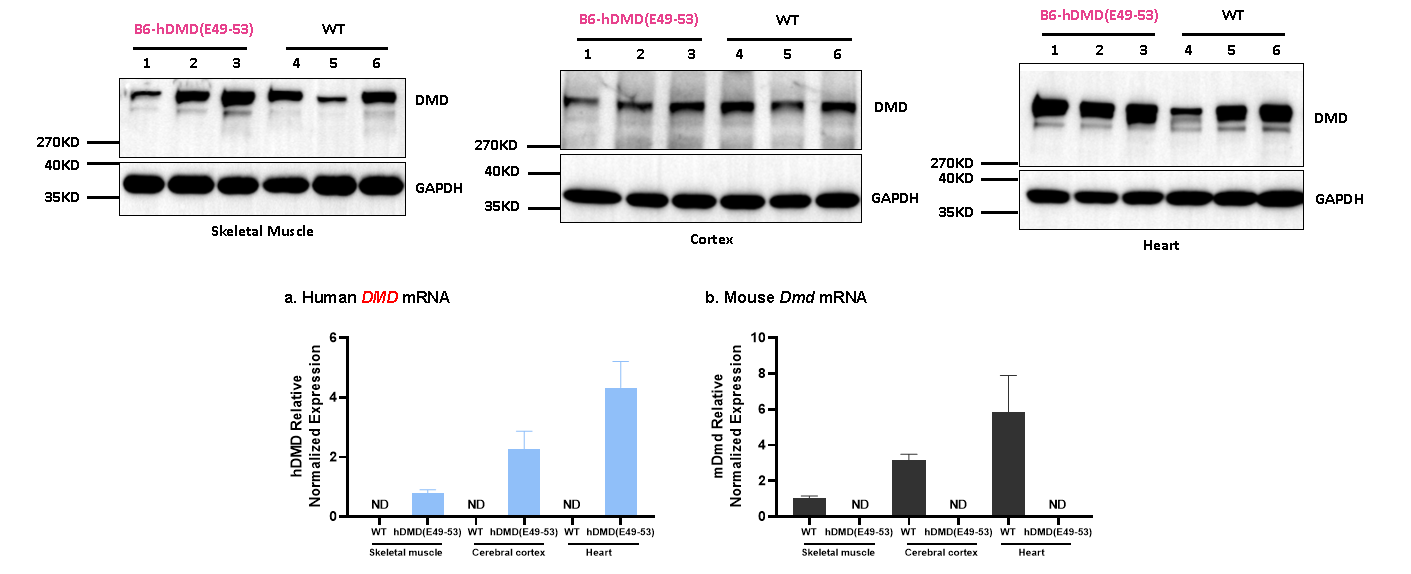
그림 8. 인간 DMD 유전자와 전장 디스트로핀(Dystrophin) 단백질의 발현에 성공한 B6-hDMD(E49-53) 마우스.
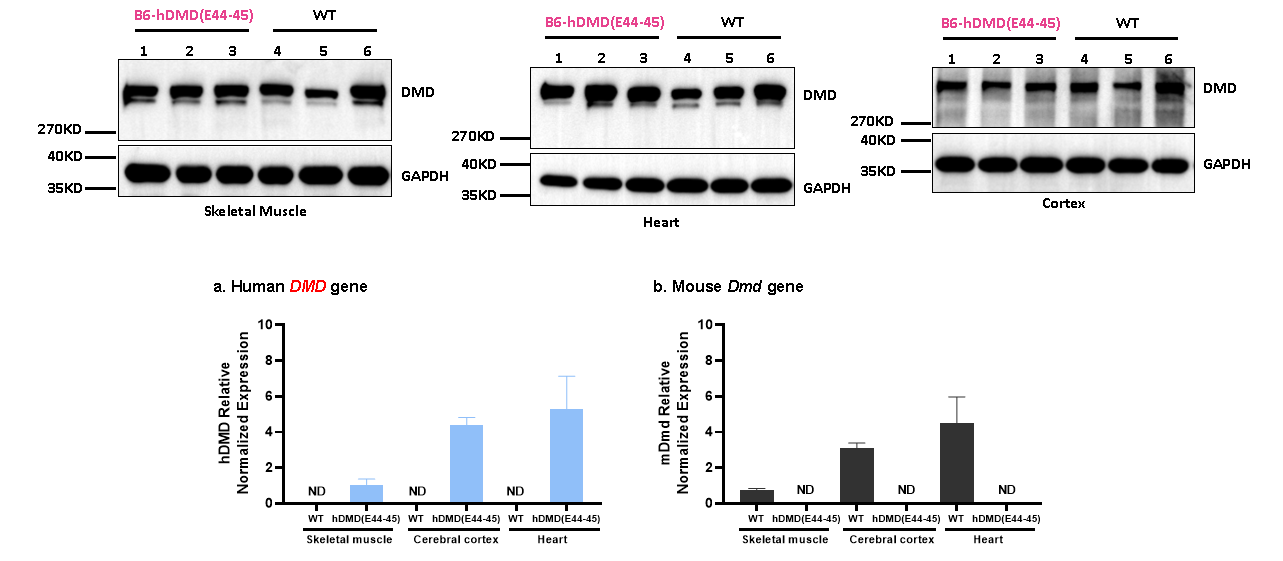
그림 9. 인간 DMD 유전자와 전장 디스트로핀(Dystrophin) 단백질의 발현에 성공한 B6-hDMD(E44-45) 마우스.
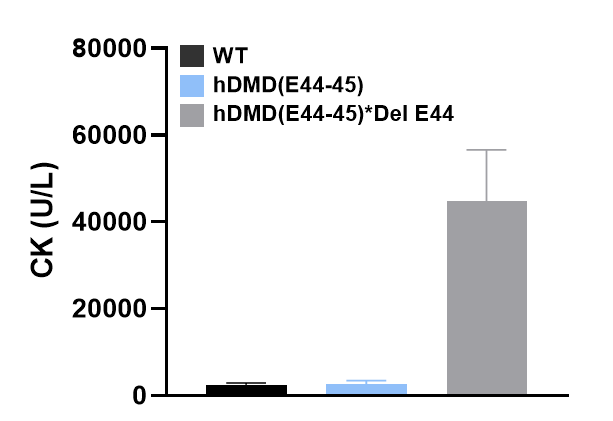
그림 10. 엑손 44 결실로 인해 기능적 장애와 심각한 근육 손상 표현형을 보이는 B6-hDMD(E44-45)*Del E44 마우스 모델.
유전자 편집 기술과 핵산 기반 치료제가 지속적으로 발전함에 따라 듀센 근이영양증(DMD)에 대한 새로운 치료 접근법이 등장하고 있습니다. 인간화 마우스 모델은 이러한 혁신을 촉진하는 데 중요한 역할을 하며 듀센 근이영양증(DMD) 환자와 그 가족에게 새로운 희망을 가져다줍니다.
HUGO-GT™ 이니셔티브: 차세대 인간화 모델 공동 개발
Cyagen은 HUGO-GT™ (Humanized Genomic Ortholog for Gene Therapy) 프로그램을 통해, 글로벌 사업 파트너들과 함께 차세대 전장 게놈 기반 인간화 모델을 개발하고 신약 개발을 가속하고자 합니다.
Cyagen 연구팀은 듀센 근이영양증(DMD) 전임상 연구에 활용할 수 있는 다양한 전장 게놈 인간화 마우스 모델을 개발하였으며, Wild-type(WT), ㅡMutant (Mut), Knockout(KO) 모델을 포함합니다. 이 모델들은 듀센 근이영양증(DMD) 환자에서 고빈도로 발생하는 exon 돌연변이 영역을 기반으로 설계되었습니다.
Cyagen은 항상 혁신을 추진하고 있으며, 연구에 맞춤화되는 HUGO-GT™ 마우스 모델을 제공해 드립니다. 원하시는 연구 모델을 못 찾을 경우, 언제든지 Cyagen에게 문의해 주세요.
Cyagen HUGO-GT™ 마우스 모델은 독점적인 TurboKnockout-Pro 기술을 기반으로 개발되었으며, 마우스 유전자의 in situ 치환을 가능하게 합니다. 이 모델은 보다 폭넓은 intervention 타겟을 포함하고 있으며, 유전자 large-fragment 융합 기술이 적용되어 있습니다. 다양한 연구 목적에 활용할 수 있는 유연한 플랫폼으로, HUGO-GT™ 프로그램은 맞춤형 변이 유도 서비스를 제공하며, 실제 생물학적 메커니즘을 더욱 정밀하게 반영하는 전임상 연구 모델을 제공합니다.
참고 문헌:
[1]Min YL, Bassel-Duby R, Olson EN. Targeted Gene Editing Correction of Duchenne Muscular Dystrophy. Annu Rev Med. 2019 Jan 27;70:239-255.
[2]Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021 Feb 18;7(1):13.
[3]Gatto F, Benemei S, Piluso G, Bello L. The complex landscape of DMD mutations: moving towards personalized medicine. Front Genet. 2024 Mar 26;15:1360224.
[4]Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barišić N, Brabec P, Lahdetie J, Walter MC, Schreiber-Katz O, Karcagi V, Garami M, Viswanathan V, Bayat F, Buccella F, Kimura E, Koeks Z, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera-Pruszczyk A, Zimowski J, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Jeannet PY, Joncourt F, Díaz-Manera J, Gallardo E, Karaduman AA, Topaloğlu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Bellgard MI, Kirschner J, Flanigan KM, Straub V, Bushby K, Verschuuren J, Aartsma-Rus A, Béroud C, Lochmüller H. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015 Apr;36(4):395-402.
[5]Horn S, Fehse B. Wie sicher ist die Gentherapie? : Zweiter Todesfall nach Therapie der Duchenne-Muskeldystrophie [How safe is gene therapy? : Second death after Duchenne therapy]. Inn Med (Heidelb). 2024 Jun;65(6):617-623. German.
[6]Bönnemann CG, Belluscio BA, Braun S, Morris C, Singh T, Muntoni F. Dystrophin Immunity after Gene Therapy for Duchenne's Muscular Dystrophy. N Engl J Med. 2023 Jun 15;388(24):2294-2296.
[7]Entrada Therapeutics. "Duchenne Muscular Dystrophy (DMD)." Entrada Therapeutics. Accessed October 31, 2023. https://www.entradatx.com/dmd.
[8]Leckie J, Zia A, Yokota T. An Updated Analysis of Exon-Skipping Applicability for Duchenne Muscular Dystrophy Using the UMD-DMD Database. Genes (Basel). 2024 Nov 20;15(11):1489.
[9]van Deutekom J, Beekman C, Bijl S, Bosgra S, van den Eijnde R, Franken D, Groenendaal B, Harquouli B, Janson A, Koevoets P, Mulder M, Muilwijk D, Peterburgska G, Querido B, Testerink J, Verheul R, de Visser P, Weij R, Aartsma-Rus A, Puoliväli J, Bragge T, O'Neill C, Datson NA. Next Generation Exon 51 Skipping Antisense Oligonucleotides for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2023 Jun;33(3):193-208.
[10]Sheri N, Yokota T. In Vivo Evaluation of Exon 51 Skipping in hDMD/Dmd-null Mice. Methods Mol Biol. 2023;2640:327-336.
[11]Marchal GA, van Putten M, Verkerk AO, Casini S, Putker K, van Amersfoorth SCM, Aartsma-Rus A, Lodder EM, Remme CA. Low human dystrophin levels prevent cardiac electrophysiological and structural remodelling in a Duchenne mouse model. Sci Rep. 2021 May 7;11(1):9779.
[12]Yavas A, Weij R, van Putten M, Kourkouta E, Beekman C, Puoliväli J, Bragge T, Ahtoniemi T, Knijnenburg J, Hoogenboom ME, Ariyurek Y, Aartsma-Rus A, van Deutekom J, Datson N. Detailed genetic and functional analysis of the hDMDdel52/mdx mouse model. PLoS One. 2020 Dec 23;15(12):e0244215.
[13]Echigoya Y, Lim KRQ, Trieu N, Bao B, Miskew Nichols B, Vila MC, Novak JS, Hara Y, Lee J, Touznik A, Mamchaoui K, Aoki Y, Takeda S, Nagaraju K, Mouly V, Maruyama R, Duddy W, Yokota T. Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol Ther. 2017 Nov 1;25(11):2561-2572.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



