

헌팅턴병(Huntington's disease, HD)은 상염색체 우성 방식으로 유전되는 드문 진행성 뇌 질환입니다. 이 질환은 뇌를 변화시키는 헌팅틴(HTT) 단백질의 결함으로 인해 발생하며, 환자들이 행동, 사고 및 무의식적인 움직임에 문제를 경험하게 한다. HTT 유전자는 헌팅틴 단백질을 암호화하며 헌팅턴병의 주요 병원성 유전자이다.
이 기사에서 우리는 HTT의 기능을 검토하고 HTT 유전자 연구에 대한 견해를 제시하면서 헌팅턴병에 대한 표적 유전자 치료에서의 역할을 탐구하고자 한다.
유전자 기본 정보
|
Species |
Human |
Mouse |
Rat |
|
Chromosome |
4 |
5 |
14 |
|
Full Length |
169,280 |
150,795 |
149,499 |
|
mRNA(nt) |
13,472 |
12,237 |
13,189 |
|
Numbers of exons |
67 |
67 |
68 |
|
Numbers of amino acids |
3142 |
3120 |
3120 |
|
Cyagen Mouse Models |
|||
|
Status |
Custom |
Catalog Models |
Live Mice |
|
Knockout (KO) |
√ |
√ |
|
|
Conditional Knockout (cKO) |
√ |
|
|
비고: √로 체크된 것은 Cyagen AI Knock-Out Mouse Model eBank에서 이용할 수 있는 해당 모델을 나타낸다.
HTT 유전자 연구 상황
HTT 유전자는 헌팅턴병(Huntington disease, HD)과 직접 관련이 있으며, 질병을 일으키는 구역은 엑손 1에 있다. 정상인의 경우 HTT 유전자의 엑손 1의 수는 35회를 넘지 않는 연속적인 CAG 반복이 있으며, Huntingtin 단백질의 폴리글루탐산을 암호화한다. 이런 CAG 반복 횟수가 35회 이상일 때, CAG 반복 횟수 증가와 함께 HD 발생률이 높아진다. 현재 유럽과 미국의 코카서스 백인들 중 HD 발병률은 0.005~0.01%로 희귀질환 범주에 속하지만 이 질병의 병원성 유전자가 단일하고 발견 시기가 빠르며, 또한 증상은 운동, 인지, 정신 등의 다중 장애에 관련되어 있기 때문에 신경퇴행성 질환 분야에 대한 심도있는 연구가 진행되고 있다. 그러나 아직 시판중인 HD 관련 치료제는 없다.
인간 HTT 단백질은 4번 염색체의 short arm에 있고 길이가 180bp이며 67개의 엑손이 있다. 헌팅턴병의 병원성 부위는 첫 번째 엑손에 있으며 HTT 유전자의 돌연변이는 대부분 haplotype A1, A2, A3의 세 가지 유형이다. HTT 단백질의 중요한 영역에는 N-말단 폴리글루타민 영역, 폴리프롤린 영역, 아미노산이 가장 많은 3개의 HEAT 영역(이 영역은 4개의 단백질에서 가장 먼저 발견됨)이 포함된다. HTT에는 Proteasome, Caspase, Calpain에 대한 인식 부위가 있다. 정상적인 상황에서 HTT는 Caspase에 의해 두 개의 세그먼트로 가수분해 될 수 있으며 두 개의 세그먼트가 결합되어 스스로 기능을 수행할 수 있다. 또한, HD의 병리학적 상황에서 HTT의 발현은 증가했을 뿐만 아니라, 그것의 유리된 N-말단 산물이 더 쉽게 응집되는 동시에 C-말단에도 일정한 독성이 생긴다.
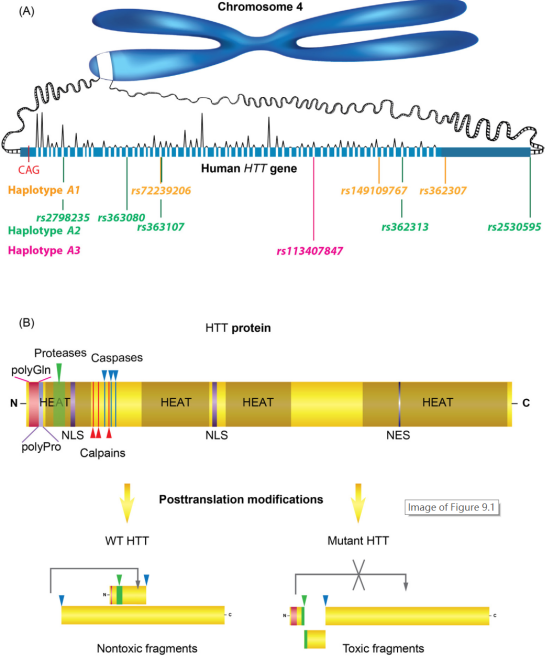
Figure 1: A) Human Huntingtin gene, HTT, and B) its protein, huntingtin (HTT), with an overview of posttranslational modifications.
ISBN: 978-0-12-805120-7
정상적인 HTT는 BDNF의 전사를 촉진하고 피질-선조체 축삭에서 BDNF의 수송을 촉진할 수 있다. BDNF가 피질과 선조체 사이의 시냅스로 방출되면 선조체에 있는 TrkB 수용체를 활성화시켜 TrkB의 세포내 섭취를 일으킨다. 세포내 섭취된 TrkB는 Dynein, Dynactin, Kinesin-1과 함께 복합체를 형성하여 Erk1/2를 활성화시켜 뉴런의 생존을 촉진한다.
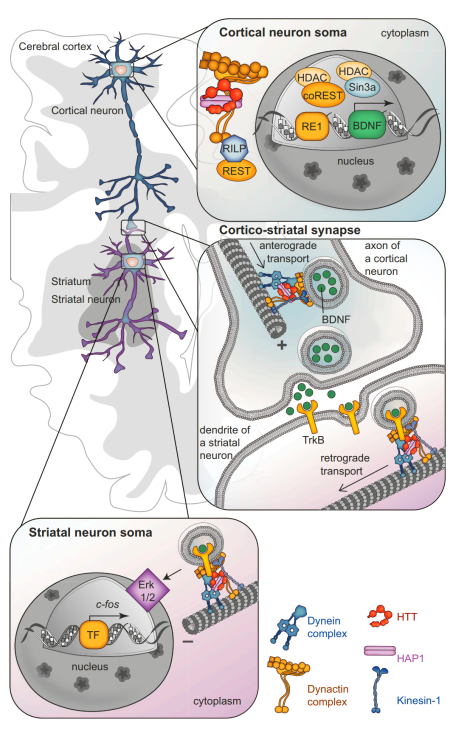
Figure 2. HTT Regulates the Function of the Cortico-Striatal Connection
DOI: 10.1016/j.neuron.2016.02.003
인간 조직에서 HTT 유전자의 발현
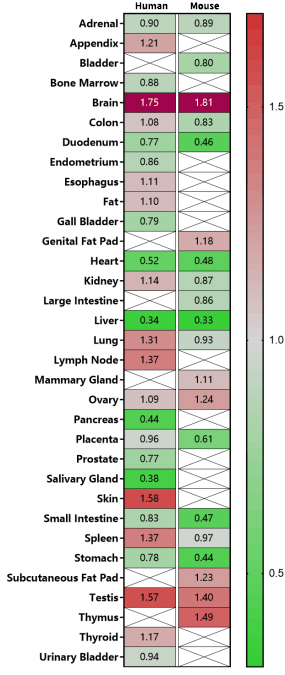
Figure 3. Expression of Human HTT and mouse Htt gene mRNA (relative expression values). This gene is undoubtedly expressed at an absolute high level in human and mouse brain tissues. HTT mRNA is also highly expressed in the testis, skin, spleen, and lungs. In addition, the expression in thymus is relatively high. (The expression information is normalized relative value rather than direct PRKM data. The comparison above is normalized within the same species; exact expression level values between mice and human are not to be compared) Data source: NCBI.
HTT 유전자 및 단백질 기능에 대한 기초 연구가 지속적으로 심화됨에 따라 핵산 약물 연구 개발과 다양한 유전자 공학 기술이 급속한 발전을 이루었다. 더욱 편리하고 안전하며 효과적인 치료제가 임상 시험에서 성공을 거두어 임상으로 나아갈 것이다.
Cyagen은 바이오 분야에 다년간 종사하여 신경질환, 특히 신경퇴행성 질환에 관한 연구에 풍부한 경험을 축적했습니다. 다양한 신경퇴행성 질환의 중요한 표적에 대해, Cyagen은 해당 유전자 편집 동물을 개발하여 연구자들이 신경퇴행성 질환의 발생 및 발달 메커니즘, 약물 선별 및 치료 효과 평가를 연구하는데 도움을 줍니다. 신경퇴행성 질환 관련 동물모델 필요한 경우 언제든지 연락해 주시기 바랍니다.
1. Saudou,Frédéric,Humbert S.The Biology of Huntingtin[J].Neuron,2016,89(5):910-926.
2. Hersch SM,Rosas HD.Biomarkers to Enable the Development of Neuroprotective Therapies for Huntington’s Disease.In:Lo DC,Hughes RE,editors.Neurobiology of Huntington's Disease:Applications to Drug Discovery.Boca Raton(FL):CRC Press/Taylor&Francis;2011.Chapter 11.PMID:21882408.
3. Ehrnhoefer DE,Wong BK,Hayden MR.Convergent pathogenic pathways in Alzheimer's and Huntington's diseases:shared targets for drug development.Nat Rev Drug Discov.2011 Oct 21;10(11):853-67.doi:10.1038/nrd3556.PMID:22015920;PMCID:PMC3206090.
4. ISBN: 978-0-12-805120-7
5. Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016 Mar 2;89(5):910-26. doi: 10.1016/j.neuron.2016.02.003. PMID: 26938440.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



