

인체의 몸에 에너지를 공급하기 위해 존재하는 글리코겐(glycogen)이 근육과 장기에 축적되어 점차 사람의 힘과 움직임을 앗아가는 질환이 있다면 어떨까요? 이것이 바로 전 세계적으로 약 40,000명 중 1명에게 영향을 미치는 희귀 유전질환, 폼페병(Pompe Disease) 환자들이 겪고 있는 현실입니다. 이러한 파괴적인 질환에 대해 더욱 효과적인 치료법을 개발하려는 연구가 계속되는 가운데, Gaa Knockout 마우스 모델은 과학 연구의 최전선에 유용한 연구 모델 중 하나로 중요한 역할을 합니다.
본문은 Gaa KO 마우스 모델이 어떻게 폼페병의 복잡한 병태 생리를 규명하고, 환자 예후를 획기적으로 개선할 수 있는 최신 치료법을 평가하는 데 활용되고 있는지 소개합니다. 본문은 신뢰할 수 있는 질환 모델을 찾는 연구자, 치료 기술 발전을 주시하는 보건의료 종사자, 혹은 폼페병으로 직접 영향을 받은 분 모두에게 앞으로의 의학 발전을 이끄는 과학적 인사이트를 제공합니다.
폼페병(Pompe Disease) 이해하기
폼페병(Pompe Disease)의 발병 기전은 에너지의 중요한 원천이 되어야 할 글리코겐이 오히려 "달콤한 부담"이 되어 체내에 축적되고, 근육과 장기를 침식합니다. 폼페병(Pompe Disease)은 제2형 당원병(Glycogen Storage Disease Type II, GSD II)으로도 알려져 있으며, 희귀한 유전성 리소좀 축적 질환(Lysosomal Storage Disorders, LSDs)입니다.[1] 이 질환은 환자의 힘을 점차 빼앗으며, 특히 영아기 및 소아기 환자에게 심장 및 호흡기계에 심각한 영향을 미칩니다.
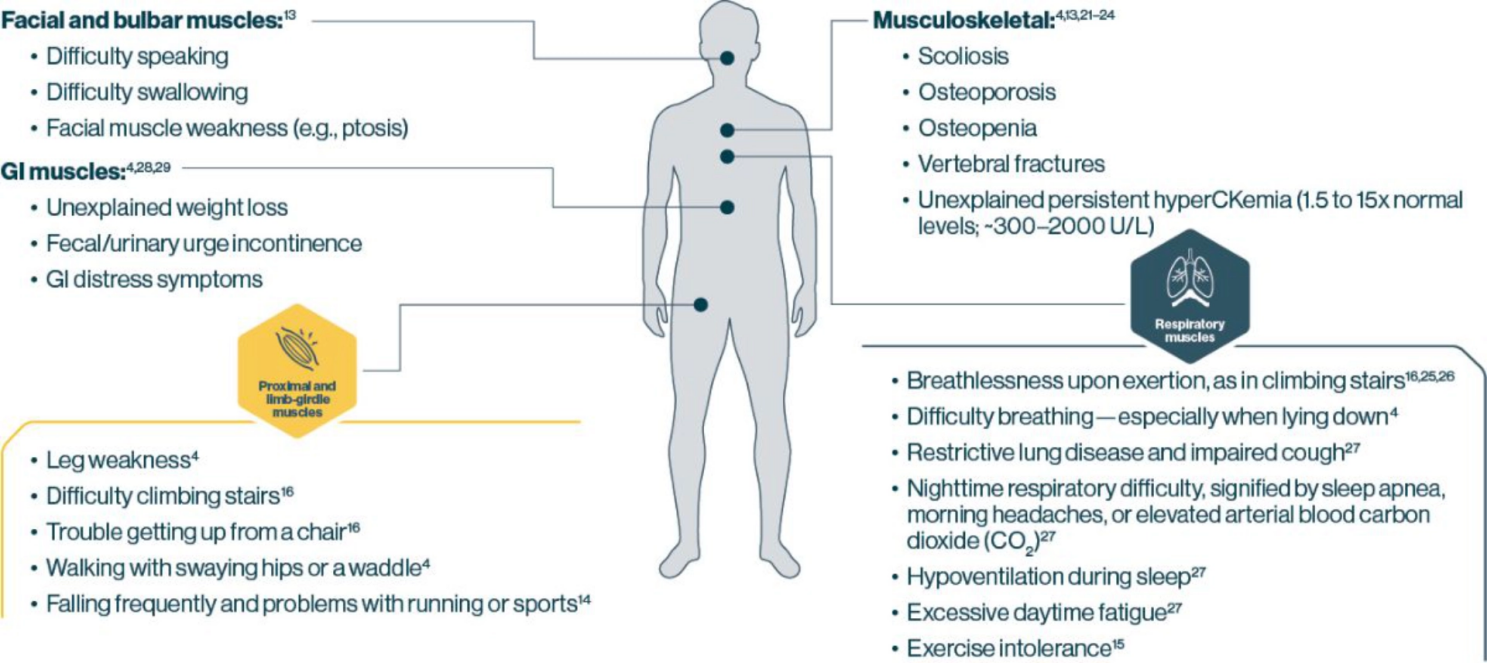
그림 1. 운동 기능 장애와 호흡 장애는 폼페병(Pompe Disease)의 가장 흔한 증상입니다. [1]
폼페병(Pompe Disease)의 비밀을 밝히다: GAA 결핍과 글리코겐의 “포위망”
폼페병(Pompe Disease)은 GAA 유전자의 돌연변이로 인해 발생하는 희귀 유전성 리소좀 축적 질환(LSD)입니다. GAA 유전자는 리소좀 내에서 글리코겐을 분해하는 데 필수적인 효소인 GAA(Acid Alpha-Glucosidase)를 인코딩합니다. GAA(Acid Alpha-Glucosidase)가 결핍되거나 기능을 하지 못할 경우:
전 세계적으로 폼페병(Pompe Disease)의 발병률은 약 40,000명 중 1명으로 추정됩니다. 발병 시기 및 질환의 중증도에 따라 크게 영아기 발병형(Infantile-Onset Pompe Disease, IOPD)과 성인 후기 발병형(Late-Onset Pompe Disease, LOPD)으로 구분됩니다. [1–2]
영아기 발병형 폼페병(IOPD)의 병세는 매우 심각하며, 대부분 환자가 생후 수개월 내에 다음과 같은 증상을 보입니다:
적절한 치료가 없으면, 영아기 발병형 폼페병(IOPD)은 조기 사망으로 이어지는 경우가 많습니다.
반면, 후기 발병형 폼페병(LOPD)은 진행 속도가 더 느리고, 일반적으로 소아기 또는 성인기에 발병합니다. 후기 발병형 폼페병(LOPD)의 주요 특징은 다음과 같습니다:
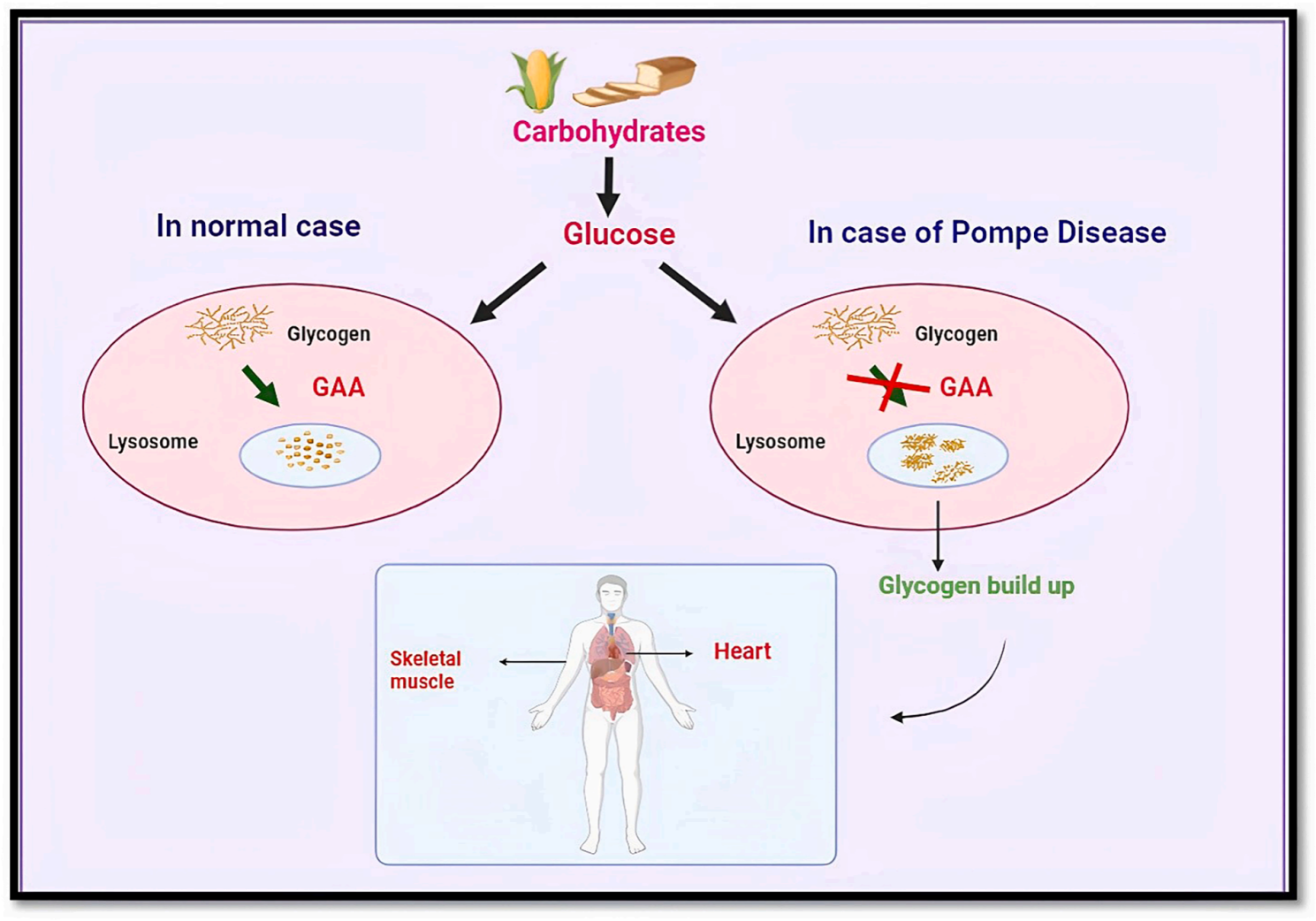
그림 2. GAA 돌연변이에 의한 폼페병(Pompe Disease)의 발병 기전. [4]
치료의 진전: 효소 대체 요법에서 유전자 치료의 서막까지
다행히도 의학 기술의 발전으로 폼페병(Pompe Disease) 치료에 있어 눈에 띄는 진전이 이루어지고 있습니다. 현재 사용 중이거나 개발 중인 주요 치료 전략은 다음과 같습니다:
효소 대체 요법(ERT)은 현재 폼페병(Pompe Disease)의 주요 치료법으로, 환자 체내에 결핍된 효소를 보충하기 위해, 재조합 인간 GAA(recombinant human GAA, rhGAA)를 정맥 주사로 투여하여 질환의 진행을 늦춥니다. 대표적인 약물로는 Alglucosidase alfa(품명: Myozyme®/Lumizyme®)가 이에 해당됩니다. [3-5]
그러나 효소 대체 요법(ERT)에는 다음과 같은 한계점이 존재합니다:
전 세계 연구자들은 더 많이 발전된 치료 접근법을 활발히 연구하고 있습니다.
이 획기적인 접근법은 AAV(Adeno-associated Virus) 등의 바이러스를 이용해 기능성 GAA 유전자를 환자 체내에 전달함으로써 "단 한 번의 투여로 장기적인 효과"를 기대합니다. 현재 폼페병(Pompe Disease)을 대상으로 한 여러 유전자 치료 임상시험이 진행 중이며, 치료 패러다임을 근본적으로 바꿀 가능성을 보여주고 있습니다.
강화된 타겟팅 기능, 연장된 반감기 또는 더 높은 활성을 갖춘 개선된 효소 제제 개발은 지속적으로 진행되고 있습니다.
기질 감소 요법 (SRT)은 글리코겐 합성을 억제함으로써 리소좀 내의 부담을 줄이는 전략입니다.
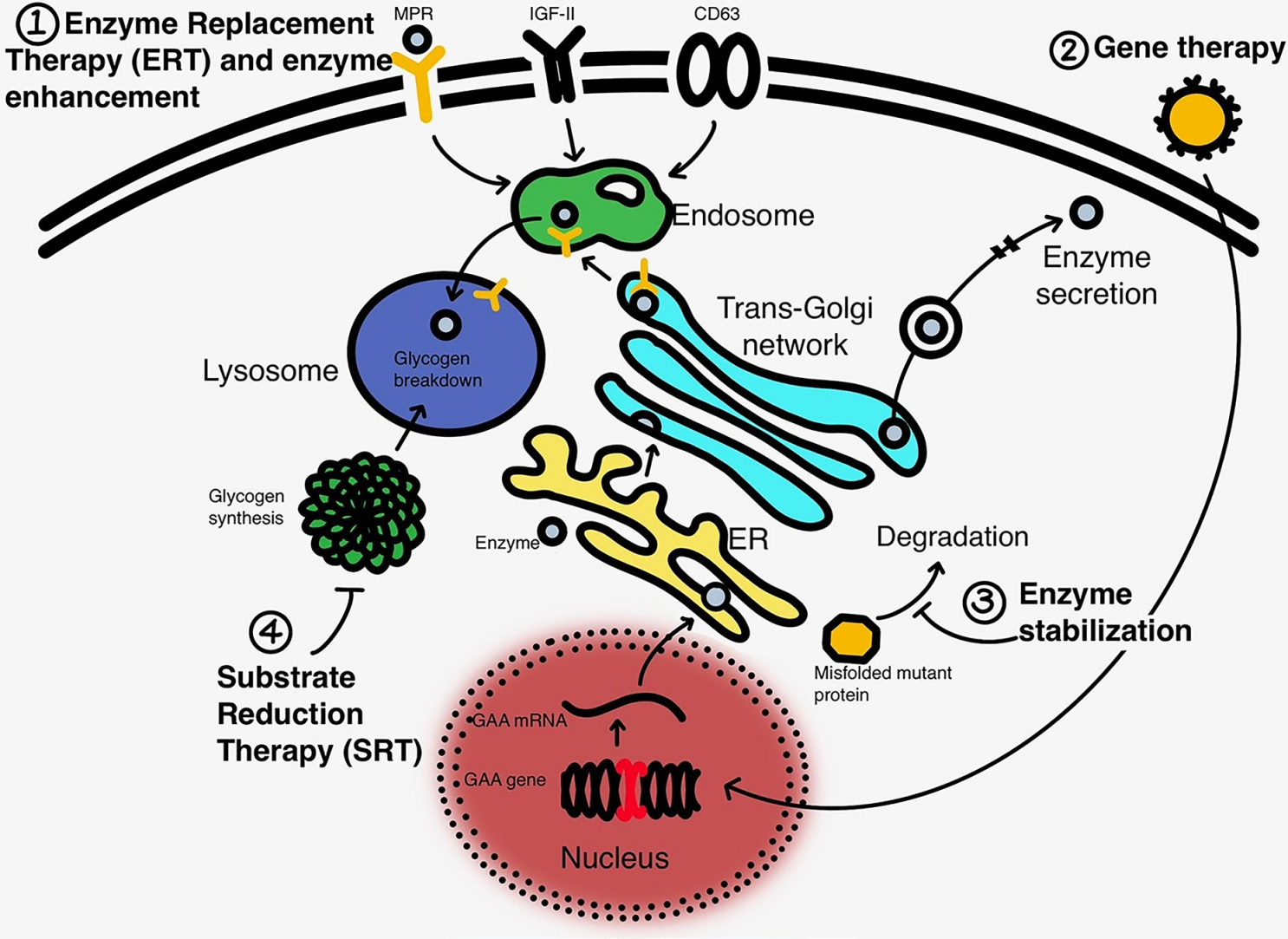
그림 3. 세포 내(간 또는 골격근)에서 다양한 폼페병(Pompe Disease) 치료법의 메커니즘을 보여주는 개념도. [11]
유용한 연구 모델: Gaa Knockout 마우스
폼페병(Pompe Disease)의 효과적인 치료 개발은 신뢰할 수 있는 전임상 모델에 기반합니다. 마우스의 Gaa 유전자는 인간의 GAA 유전자와 높은 상동성(homology)을 가지기 때문에, 마우스 모델은 폼페병(Pompe Disease) 연구에서 중요한 역할을 합니다. Gaa Knockout(KO) 마우스는 다음과 같은 인간 폼페병(Pompe Disease)의 주요 병리적 특징을 잘 재현합니다:
이러한 특성 덕분에 Gaa Knockout(KO) 마우스는 다음과 같은 연구에 필수적인 모델로 활용됩니다:
따라서 Gaa Knockout(KO) 마우스는 질환 발병 기전 규명, 약물 후보 물질 스크리닝, 치료 반응 평가, 그리고 새로운 유전자 치료법의 안전성과 효능을 검증하는 데 이상적인 모델로 활용됩니다.
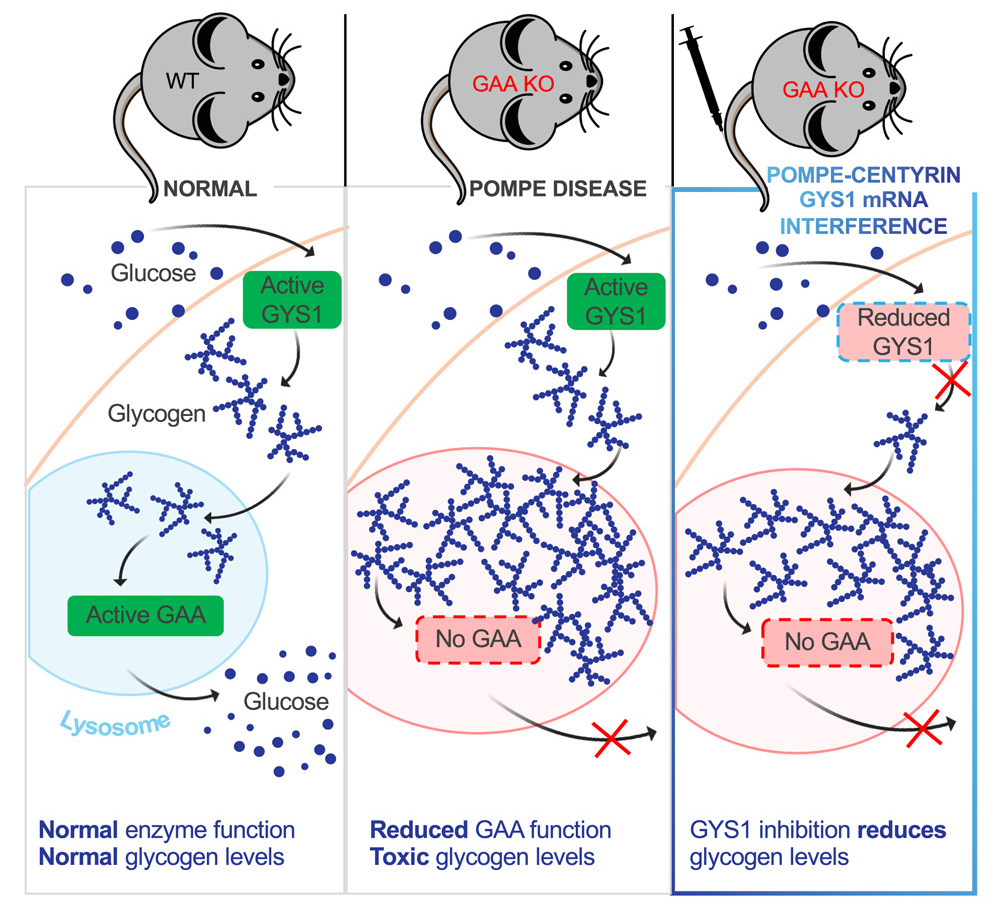
그림 4. Gaa KO 마우스를 활용한 연구 사례: 폼페병(Pompe Disease)에 대한 RNA 간섭 치료법의 전임상 평가. [15]
Cyagen의 Gaa KO 마우스: 신뢰할 수 있는 데이터 기반의 정밀 질환 모델링
폼페병(Pompe Disease) 연구의 시급한 니즈에 대응하기 위해, Cyagen은 Gaa Knockout(KO) 마우스 모델을 개발하였습니다 (제품 번호: C001702).
초기 데이터(Preliminary data)에 따르면, 이 마우스는 폼페병(Pompe Disease)의 주요 특징을 잘 재현하며, 구체적으로 다음과 같은 병리적 특징을 보입니다.
아래는 Gaa Knockout(KO) 마우스 모델에 대한 광범위한 특성 분석 중 일부 검증 데이터입니다. (전체 내용은 계통 데이터시트에서 확인할 수 있습니다.)
Gaa KO 마우스는 악력 테스트(Grip strength test)에서 wild-type(WT) 대조군에 비해 유의미하게 낮은 근력을 보이며, 이는 폼페병(Pompe Disease) 환자에서 나타나는 근력 약화를 반영합니다.
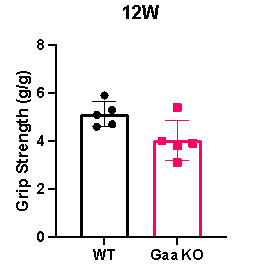
그림 5. 12주령 Homozygous 암컷 Gaa KO 마우스와 wild-type(WT) 대조군(n = 5)의 근력 테스트 결과.
12주령의 Gaa KO 마우스는 wild-type(WT) 마우스에 비해 보다 현저한 체중 증가를 보이며, 이는 질환에서 관찰되는 대사 이상과 연관이 있습니다.
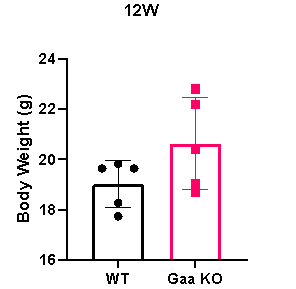
그림 6. 12주령 Homozygous 암컷 Gaa KO 마우스와 wild-type(WT) 대조군(n = 5)의 체중 데이터.
Gaa KO 마우스는 주요 조직에서 wild-type(WT) 대조군에 비해 현저히 높은 글리코겐 축적을 보입니다.
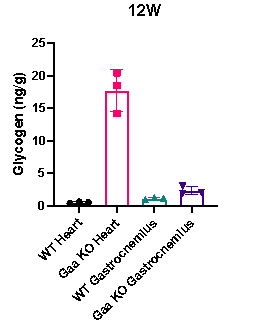
그림 7. 12주령 Homozygous 암컷 Gaa KO 마우스와 wild-type(WT) 대조군(n = 3)의 심장 및 장딴지근 내 글리코겐의 함량.
Gaa KO 마우스는 wild-type(WT) 대조군과 비교했을 때 심장 및 장딴지근에서 GAA 효소 활성이 현저히 저하되어 있습니다.

그림 8. 12주령 Homozygous 암컷 Gaa KO 마우스와 wild-type(WT) 대조군(n = 3)의 심장과 장딴지근에서의 GAA 효소 활성.
결론: 안정적인 모델로 폼페병(Pompe Disease) 연구를 추진합나다
Gaa Knockout(KO) 마우스 모델 (제품 번호: C001702) 은 다음과 같은 폼페병(Pompe disease)의 주요 병리적 특징을 성공적으로 재현합니다.
Gaa KO 마우스 모델은 안정적인 표현형과 신뢰도 높은 실험 데이터를 바탕으로, 다음과 같은 연구에 적용됩니다.
Cyagen은 고품질 Gaa KO 마우스 모델이 폼페병(Pompe disease) 연구를 진전시키고, 혁신적인 치료법 개발을 가속하여 이 희귀 유전질환으로 고통받는 전 세계 환자들에게 희망을 줄 수 있기를 바랍니다.
|
제품 번호 |
모델명 |
품종 계통 |
응용 분야 |
|
C001507 |
C57BL/6JCya |
Atherosclerosis, Hypercholesterolemia, Metabolic Dysfunction-Associated Steatohepatitis (MASH) |
|
|
C001067 |
APOE |
C57BL/6NCya |
Atherosclerosis |
|
C001291 |
B6-db/db |
C57BL/6JCya |
High Blood Sugar and Obesity |
|
C001392 |
C57BL/6JCya |
Familial Hypercholesterolemia |
|
|
C001368 |
C57BL/6JCya |
Type 2 Diabetes and Obesity |
|
|
C001232 |
C57BL/6JCya |
Hyperuricemia |
|
|
C001267 |
C57BL/6NCya |
Copper Metabolism Disorder, Wilson's Disease |
|
|
C001265 |
C57BL/6NCya |
Primary Ciliary Dyskinesia |
|
|
C001266 |
C57BL/6NCya |
Klinefelter Syndrome |
|
|
C001273 |
C57BL/6NCya |
Phenylketonuria Type 1 |
|
|
C001383 |
Alb-Cre/LSL-hLPA |
C57BL/6NCya |
Cardiovascular Targets |
|
C001421 |
C57BL/6NCya |
Metabolic Targets |
|
|
C001400 |
C57BL/6JCya |
Metabolic Targets |
|
|
C001493 |
FVB |
Diseases Related to Blood-Brain Barrier Permeability |
|
|
C001532 |
C57BL/6JCya |
Hereditary Angioedema(HAE) |
|
|
C001549 |
C57BL/6NCya |
Research on diet-induced obesity, diabetes, inflammation, fatty liver, and other metabolic diseases; drug development, screening, and preclinical efficacy evaluation for obesity. |
|
|
C001553 |
C57BL/6NCya |
Familial hypercholesterolemia (FH); atherosclerotic cardiovascular disease (ASCVD); other cardiovascular diseases (CVD). |
|
|
C001560 |
C57BL/6JCya |
Phenylketonuria (PKU) |
|
|
I001220 |
C57BL/6Cya |
Research on PCSK9-targeted drug development; studies on metabolic diseases such as hyperlipidemia, stroke, coronary heart disease, and familial hypercholesterolemia (FH). |
|
|
I001223 |
C57BL/6NCya |
Fabry Disease (FD) |
|
|
C001583 |
FVB/NJCya |
Propionic Acidemia (PA) |
|
|
C001590 |
FVB/NJCya |
Progressive Familial Intrahepatic Cholestasis Type 3 (PFIC3) |
|
|
C001594 |
C57BL/6JCya |
Glutaric aciduria type I (GA1) |
|
|
C001600 |
C57BL/6NCya; C57BL/6JCya |
Type 2 Diabetes, Obesity, and Metabolic Disorders Associated with Improper Fat Distribution and Storage |
|
|
C001601 |
C57BL/6NCya; C57BL/6JCya |
Type 2 Diabetes and Obesity |
|
|
C001591 |
C57BL/6NCya; C57BL/6JCya |
Familial hypercholesterolemia (FH); atherosclerotic cardiovascular disease (ASCVD); other cardiovascular diseases (CVD) |
|
|
C001609 |
C57BL/6JCya |
Hypertrophic Cardiomyopathy (HCM) and Dilated Cardiomyopathy (DCM) |
|
|
I001121 |
C57BL/6JCya |
Research on emphysema and chronic obstructive pulmonary disease (COPD), cirrhosis, and hepatocellular carcinoma |
|
|
I001225 |
C57BL/6NCya; C57BL/6JCya |
Autosomal Dominant Polycystic Kidney Disease (ADPKD) and Renal Tubular Biology |
|
|
C001702 |
C57BL/6JCya |
Glycogen Storage Disease Type II (Pompe disease), lysosomal glycogen metabolism |
|
|
C001703 |
C57BL/6JCya |
Primary Hyperoxaluria, glyoxylate metabolism regulation |
참고 문헌:
[1]Sanofi. (2023, February 24). Pompe - Sanofi campus. Retrieved from https://www.campus.sanofi/bh/science/rare-diseases/cutting-edge-science/2023/ar/pompe
[2]Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471-86.
[3]Schoser B, Roberts M, Byrne BJ, Sitaraman S, Jiang H, Laforêt P, Toscano A, Castelli J, Díaz-Manera J, Goldman M, van der Ploeg AT, Bratkovic D, Kuchipudi S, Mozaffar T, Kishnani PS; PROPEL Study Group. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): an international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021 Dec;20(12):1027-1037.
[4]Singh, A., Debnath, R., Saini, A., Seni, K., Sharma, A., Bisht, D. S., Chawla, V., & Chawla, P. A. (2024, June). Cipaglucosidase alfa-atga: Unveiling new horizons in Pompe disease therapy. Health Sciences Review, 11, 100160. Retrieved from https://www.sciencedirect.com/science/article/pii/S2772632024000138
[5]Diaz-Manera J, Kishnani PS, Kushlaf H, Ladha S, Mozaffar T, Straub V, Toscano A, van der Ploeg AT, Berger KI, Clemens PR, Chien YH, Day JW, Illarioshkin S, Roberts M, Attarian S, Borges JL, Bouhour F, Choi YC, Erdem-Ozdamar S, Goker-Alpan O, Kostera-Pruszczyk A, Haack KA, Hug C, Huynh-Ba O, Johnson J, Thibault N, Zhou T, Dimachkie MM, Schoser B; COMET Investigator Group. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021 Dec;20(12):1012-1026.
[6]Bolano-Diaz C, Diaz-Manera J. Therapeutic Options for the Management of Pompe Disease: Current Challenges and Clinical Evidence in Therapeutics and Clinical Risk Management. Ther Clin Risk Manag. 2022 Dec 13;18:1099-1115.
[7]Koeberl DD, Koch RL, Lim JA, Brooks ED, Arnson BD, Sun B, Kishnani PS. Gene therapy for glycogen storage diseases. J Inherit Metab Dis. 2024 Jan;47(1):93-118.
[8]Bond JE, Kishnani PS, Koeberl DD. Immunomodulatory, liver depot gene therapy for Pompe disease. Cell Immunol. 2019 Aug;342:103737.
[9]Sawada T, Kido J, Nakamura K. Newborn Screening for Pompe Disease. Int J Neonatal Screen. 2020 Apr 5;6(2):31.
[10]Salabarria SM, Nair J, Clement N, Smith BK, Raben N, Fuller DD, Byrne BJ, Corti M. Advancements in AAV-mediated Gene Therapy for Pompe Disease. J Neuromuscul Dis. 2020;7(1):15-31.
[11]Stevens D, Milani-Nejad S, Mozaffar T. Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Curr Treat Options Neurol. 2022 Nov;24(11):573-588.
[12]Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L, LaMarca M, King C, Ward J, Sauer B, Plotz P. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998 Jul 24;273(30):19086-92.
[13]Raben N, Nagaraju K, Lee E, Plotz P. Modulation of disease severity in mice with targeted disruption of the acid alpha-glucosidase gene. Neuromuscul Disord. 2000 Jun;10(4-5):283-91.
[14]Fusco AF, McCall AL, Dhindsa JS, Zheng L, Bailey A, Kahn AF, ElMallah MK. The Respiratory Phenotype of Pompe Disease Mouse Models. Int J Mol Sci. 2020 Mar 24;21(6):2256.
[15]Holt BD, Elliott SJ, Meyer R, Reyes D, O'Neil K, Druzina Z, Kulkarni S, Thurberg BL, Nadler SG, Pederson BA. A novel CD71 Centyrin:Gys1 siRNA conjugate reduces glycogen synthesis and glycogen levels in a mouse model of Pompe disease. Mol Ther. 2025 Jan 8;33(1):235-248.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



