

A rare disease is defined by the European Union as a condition that affects fewer than 1 in 2,000 people within the general population. Currently, there are over 6,000 known rare diseases and newly discovered conditions are being described through medical literature on a regular basis. However, these rare diseases still affect a significant portion of the population, as roughly 1 in 17 people will be affected by a rare disease. The majority of rare diseases currently have no effective treatment [1].
Cyagen assists in the research of rare diseases and provides a wide range of mouse and rat model development services. This article focuses on the gene INPP5E and its related rare diseases, aiming to help interested researchers continue to build on scientific literature and knowledge.
Overview of INPP5E Gene
INPP5E (Inositol Polyphosphate-5-Phosphatase E) is a Protein Coding gene. Diseases associated with INPP5E include Joubert Syndrome 1 and Mental Retardation, Truncal Obesity, Retinal Dystrophy, and Micropenis (MORM) Syndrome.
The protein encoded by this gene is an inositol 1,4,5-trisphosphate (InsP3) 5-phosphatase. InsP3 5-phosphatases hydrolyze Ins(1,4,5)P3, which mobilizes intracellular calcium and acts as a second messenger mediating cell responses to various stimulation. Studies of the mouse counterpart suggest that this protein may hydrolyze phosphatidylinositol 3,4,5-trisphosphate and phosphatidylinositol 3,5-bisphosphate on the cytoplasmic Golgi membrane and thereby regulate Golgi-vesicular trafficking. Mutations in this gene cause Joubert syndrome; a clinically and genetically heterogeneous group of disorders characterized by midbrain-hindbrain malformation and various associated ciliopathies that include retinal dystrophy, nephronophthisis, liver fibrosis, and polydactyly. Alternative splicing results in multiple transcript variants encoding different isoforms. [2]
Molecular function - Inpp5E in primary cilia assembly
Previous studies have shown INPP5E has been associated with ciliary dysfunction. [3] The primary cilium (PC) is a plasma membrane-derived structure of great importance for cell and organismal physiology. Indeed, deficiencies in two 5′ PIP phosphatases, Inpp5E and Ocrl1, are clearly linked to ciliopathies like Joubert/MORM syndromes, or ciliopathy-associated diseases like Lowe syndrome.
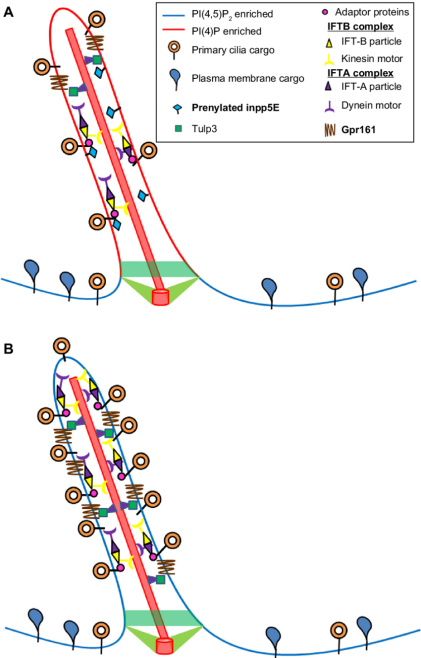
Figure 1. Ciliary role of Inpp5e. Notes: (A) Inpp5e converts the Pi(4,5)P2 into Pi(4)P by functioning as a 5′ phosphatase in the cilium. It has been shown to interact with the adaptor protein Tulp3 to traffic ciliary cargo. (B) in the absence of a functional Inpp5e, primary cilium accumulates Pi(4,5)P2, and the cilium sequesters intraflagellar transport particles and the adaptor protein. As a result, ciliary disassembly is affected. [3]
Rare Diseases Associated with the INPP5E Gene
Joubert Syndrome (JBTS)
Joubert syndrome is a neurodevelopmental disorder, characterized by malformation of the mid and hindbrain leading to the pathognomonic molar tooth appearance of the brainstem and cerebellum on axial MRI. Core clinical manifestations include hypotonia, tachypnea/apnea, ataxia, ocular motor apraxia, and developmental delay of varying degrees. In addition, a subset of patients has retinal dystrophy, chorioretinal colobomas, hepatorenal fibrocystic disease, and polydactyly. Joubert syndrome exhibits genetic heterogeneity, with mutations identified in more than 30 genes, including INPP5E, a gene encoding inositol polyphosphate 5-phosphatase E, which is important in the development and stability of the primary cilium. [4]
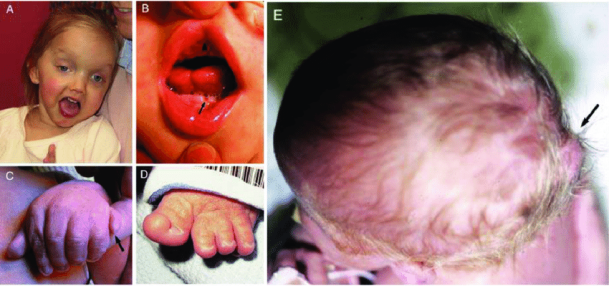
Figure 2. Clinical features in Joubert syndrome. (A) Facial features in a girl with Joubert syndrome (COACH syndrome) at age 27 months showing broad forehead, arched eyebrows, strabismus, eyelid ptosis (on right eye), and open mouth configuration indicating reduced facial tone. (B) Oral findings in a child with oral-facial-digital syndrome-like features of Joubert syndrome showing midline upper lip cleft (arrowhead), midline groove of tongue, and bumps of the lower alveolar ridge (arrow). (C) Left hand of an infant with Joubert syndrome and postaxial polydactyly (arrow). (D) Left foot of an infant with Joubert syndrome and preaxial polydactyly of the hallux. (E) View from above of an infant with a small occipital encephalocele with protrusion of the occiput of the skull (arrow). [5]
Hepatorenal fibrocystic diseases (HRFCDs)
Hepatorenal fibrocystic disorders (HRFCDs) are characterized by developmental portobiliary and renal fibrocystic abnormalities such as polycystic kidneys and congenital hepatic fibrosis. HRFCDs belong to a larger group of diseases called ciliopathies that are caused by structural or functional defects in the primary cilium. Ciliopathies are an expanding group of diseases that are clinically and genetically very heterogeneous and can manifest as a congenital developmental syndrome or as progressive single organ dysfunction. Autosomal recessive and dominant polycystic kidney diseases (ARPKD and ADPKD respectively) are the most common forms of HRFCDs in humans. Kidney and liver lesions also variably occur in other syndromic ciliopathies like Joubert, Meckel, Bardet-Biedl and Jeune syndrome.[6] Kati J. Dillard and colleagues found that a splice site variant in INPP5E causes diffuse cystic renal dysplasia and hepatic fibrosis in dogs.
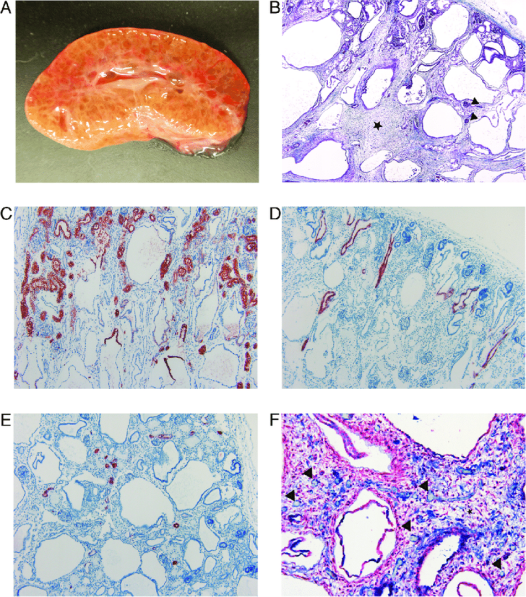
Figure 3. Macroscopic and histological changes of the affected kidneys.(A) Photograph of the cross-section of an affected kidney. Cortex and medulla are poorly demarcated and the parenchyma is nearly diffusely cystic. (B) Masson trichrome stain. There are immature glomeruli (black arrowheads) and normal appearing tubules in the cortex. The demarcation between the cortex and medulla is poorly defined. The tubules in the medulla are dilated and there is an increased amount of loose connective tissue (black star) between the cystic tubules (4X). (C) IHC staining for AQP-1 as a marker for epithelial cells (red) of the convoluted and straight part of the proximal tubule and the thin descending limb of Henle’s loop. The convoluted portions of proximal tubules appeared morphologically normal. In the upper part of the cysts, the epithelium was positively stained indicating their origin as the straight portion of the proximal tubule and thin descending limb of Henle’s loop. The lower part of the cysts was not stained (10X). (D) IHC staining for AQP-2 as a marker for epithelial cells (red) of the collecting ducts. The collecting ducts were mainly normal. There were a few slightly dilated collecting ducts in the cortex (10X). (E) IHC staining for calbindin-D28K as a marker for epithelial cells (red) of the distal convoluted tubule. The distal convoluted tubules were morphologically normal (10X). (F) IHC double staining for α-SMA for mesenchymal cells (red) and vWF for endothelial cells (blue). The connective tissue (black star) contains scattered capillaries (black arrowheads). Only a very few capillaries are in close proximity to the tubules (20X). [7]
This research explores the phenotypic difference between humans and Norwich Terriers with INPP5E variants.The variant identified in their study resides in the IPPc domain of dog INPP5E similar to the human JBTS1 patients, but splice site variants have not been reported in humans so far. Maybe in the future there will be additional discoveries related to the splice site variations among species.
To Be Continued: Mouse Models of Inpp5e
Visit the next article below, in which we provide details on the applications of mouse models used for Inpp5e gene studies and new research progress.
>>Learn more: Inpp5e Mice and New Research Progress
References:
[1] European Commission-European Commission.(2020).Rare diseases.[online] Available at:https://ec.europa.eu/info/research-and-innovation/research-area/health/rare-diseases_en [Accessed 27 Jan.2020].
[2] https://www.genecards.org/cgi-bin/carddisp.pl?gene=INPP5E&keywords=INPP5E
[3] Madhivanan K,Ramadesika S, Aguilar R C. Role of Ocrl1 and Inpp5E in primary cilia assembly and maintenance: a phosphatidylinositol phosphatase relay system?[J]. Research and Reports in Biology, 2016, 7: 15-29.
[4] https://medlineplus.gov/genetics/condition/joubert-syndrome/
[5] Parisi M A.Clinical and molecular features of Joubert syndrome and related disorders[C]//American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Hoboken:Wiley Subscription Services, Inc.,A Wiley Company,2009,151(4): 326-340.
[6] Gunay‐Aygun, Meral. "Liver and kidney disease in ciliopathies." American Journal of Medical Genetics Part C: Seminars in Medical Genetics. Vol.151.No. 4.Hoboken: Wiley Subscription Services,Inc., A Wiley Company, 2009.
[7] Dillard K J, Hytönen M K, Fischer D, et al. A splice site variant in INPP5E causes diffuse cystic renal dysplasia and hepatic fibrosis in dogs[J]. PloS one, 2018, 13(9): e0204073.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



