

Genes play a highly significant role in the pathogenesis of eye diseases, some of which are highly prevalent in children and adults. Over 60% of cases of childhood blindness are caused by genetic factors (congenital glaucoma, ocular malformations, atrophy of the optic nerve, and retinitis pigmentosa). In adults, genetic factors can also be associated with serious eye diseases, including glaucoma and macular degeneration.
Reviewing Cyagen's experience in rodent model generation over the past 15 years, we have generated many mouse models for genetic variants associated with eye diseases. For example, Cep290 knockout (KO) mice were generated for the study of Leber congenital amaurosis 10 (LCA10). Not only that, but Cyagen continues to develop humanized mouse models that are more beneficial to disease research, including the study of genetically-based eye diseases. In the following content, we will introduce some of the ophthalmologically relevant humanized mouse models for eye disease research.
Application of Humanized Mice in Ophthalmic Diseases
To better understand human health and disease, researchers create a wide variety of mouse models that carry human DNA. Such humanized mice are designed to have greater physiological relevance than their classic transgenic counterparts, which includes maintaining the correct genomic context of a gene of interest to preserve physiological expression levels and correct spatiotemporal expression patterns. Where non-coding human sequences are incorporated, human-specific regulation and human gene splice isoforms may be maintained. In addition, the translated protein is expected to display the unique biochemical properties of the human analog, including potentially unique deleterious properties when mutated. [1]
Mouse models on fixed genetic backgrounds may be useful for understanding the function of the gene in the retina, the pathological mechanisms induced by its disruption, and for testing experimental approaches to treat the corresponding human ocular diseases.
Humanized Knock-In Mouse Model for Leber Congenital Amaurosis (LCA)
The murine Cep290 gene contains 53 exons (one less than in humans) and shares a high homology at DNA (86%) and protein (87% homology and 94% similarity) levels with human CEP290 (Figure 1A). In order to generate a humanized knock-in mouse model that would mimic the genotype and phenotype associated with the deep-intronic LCA-causing mutation, a recombination strategy was designed that consisted of the replacement of mouse exons 25 (mE25) and 26 (mE26), as well as intron 25 (mI25), by the human counterparts (hE26, hI26/hI26mut and hE27) (Figure 1B). All recombination events were validated by PCR and Southern blot analysis (data not shown). The isolated positive ES-cell clones were injected in blastocysts that were implanted in pseudo-pregnant females. Highly chimeric males were bred with Cre deleter females to excise the NEO cassette. The resulting heterozygous mice were used to obtain homozygous animals, which were viable and fertile. Subsequent breedings were done between homozygous animals.
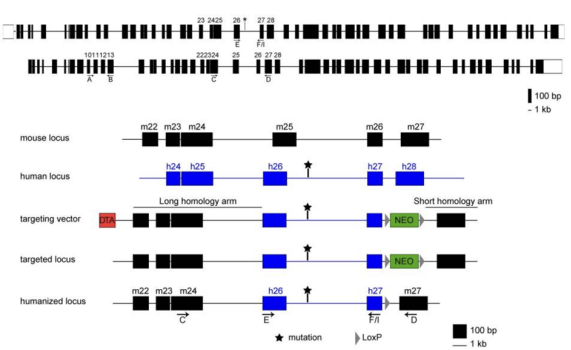
Figure. 1 Generation of the Cep290 humanized mouse models. (A) Structure of the CEP290 gene in human and mouse (drawn to scale). (B) Two mouse models were generated by introducing human exons 26 and 27 and intron 26, either with or without the LCA-causing mutation (depicted with *), to the mouse Cep290 gene. Human and mouse loci, targeting vector and recombinant locus are depicted. Arrows and letters indicate the position of the oligonucleotides [2] .
In this study, even though the humanization of mouse Cep290 was not compromising the gene expression in general, the splicing machinery of the mouse appeared not to be able to recognize the human splice acceptor site or the splice donor site that is generated by the intronic CEP290 mutation, suggesting differential recognition of splice sites in humans vs. mice. In addition, the substitution of the murine intron by the human one generated another cryptic exon, robustly expressed in the mouse retina, but not found in human retinas nor in fibroblasts. Their data suggests species-specific differences in the splicing machinery between mice and humans, indicating that mice may not always represent suitable models to study the pathogenic mechanisms underlying genetic variants that affect splicing.
However, others have successfully used humanized knock-in mouse models to mimic the pathophysiological mechanisms underlying splice site mutations for several inherited diseases in the past.
Jordan T. Gladman et. al. generated a new model of SMA using the endogenous mouse Smn gene and homologous recombination to insert the C>T SMN2 alteration into exon 7 [3]. Using comparative genomics and an in vivo splicing assay, they demonstrate that the mouse Smn and human SMN genes are regulated at exon 7 by many of the same pre-mRNA splicing elements. Furthermore, when the C>T alteration in exon 7 of the SMN2 gene is engineered into the mouse Smn gene, it increases exon 7 pre-mRNA skipping. By using homologous recombination, the modified Smn C>T allele is under the endogenous Smn promoter and in the correct genomic context. Biochemical, histological and behavioral analysis of the resultant mice are consistent with a mild adult onset form of SMA, including reduced hindlimb grip strength, decreased locomotive activity, and hypertrophic skeletal muscle fibers as seen in some Kugelberg–Welander SMA patients. The lifespan of mice is extended and allows treatment of the disease at later developmental time points, surpassing the lifespans of other SMA mouse models. This model thus has the benefit of adding to the spectrum of SMA animal models available and allows for the testing for modifiers of disease phenotype and development of therapies that directly affect exon 7 splicing.
Mouse models still play an important role in the genetic study of splicing mutations. However, for some unique splice mutations, suitable animal models still need to be found, and ethical issues may need to be faced.
What Can Cyagen Do?
Over the past year, Cyagen has focused on building an ophthalmic gene therapy platform. We aim to provide a one-stop solution for eye diseases: from generation of ophthalmic disease models to injection of AAV, and evaluation of in vitro and in vivo pharmacodynamics.
To Be Continued:
Visit the next article below, in which we provide details on the applications of mouse models used for Autosomal Dominant Retinitis Pigmentosa (ADRP).
>>Learn more: Autosomal Dominant Retinitis Pigmentosa (ADRP) & ADRP Mouse Model
[1] Zhu F, Nair RR, Fisher EMC, Cunningham TJ. Humanising the mouse genome piece by piece. Nat Commun. 2019 Apr 23;10(1):1845. doi: 10.1038/s41467-019-09716-7. PMID: 31015419; PMCID: PMC6478830.
[2] Garanto A, van Beersum SE, Peters TA, Roepman R, Cremers FP, Collin RW. Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for Leber congenital amaurosis. PLoS One. 2013 Nov 6;8(11):e79369. doi: 10.1371/journal.pone.0079369. PMID: 24223178; PMCID: PMC3819269.
[3] Gladman JT, Bebee TW, Edwards C, Wang X, Sahenk Z, Rich MM, Chandler DS. A humanized Smn gene containing the SMN2 nucleotide alteration in exon 7 mimics SMN2 splicing and the SMA disease phenotype. Hum Mol Genet. 2010 Nov 1;19(21):4239-52. doi: 10.1093/hmg/ddq343. Epub 2010 Aug 12. PMID: 20705738; PMCID: PMC2951869.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



