

Epidemiological Studies of ALS
Epidemiological studies have found that the incidence of ALS is 1.9/105, the prevalence is 4.5/105, and the average age of onset is 55 years (Incidence: proportion of new patients per year; Prevalence: ratio of existing patients to total population).
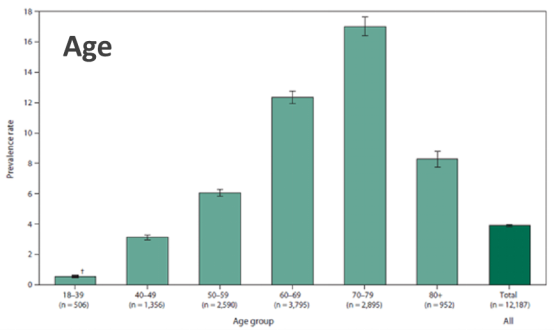
In addition, the data show that the incidence rate in Europe and the United States is higher, at 2.6/105, and the prevalence rate is 7-9/105. It is more common in men than women, and more common in whites, among whom Anglo-Saxons and Germanics have significantly higher rates than Latinos.
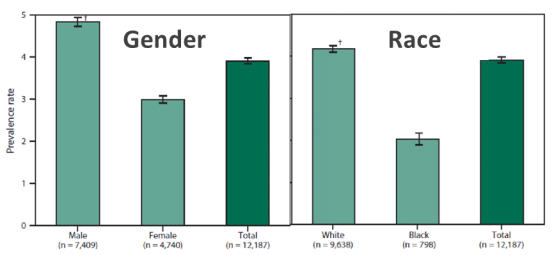
(Source: Centers for Disease Control and Prevention)
In contrast, the incidence of the yellow race is lower, 0.8/105 in East Asia, and 0.7/105 in South Asia. Because of this, Europe and the United States have invested heavily in the research of this disease, accounting for more than 80% of the world's projects on ALS.
Motor Neuron Disease
ALS is a motor neuron disease, which is a group of neurological disorders that selectively affect motor neurons with secondary damage to muscles. In a normal body, there is a motor nervous system that works in an orderly manner, as shown below:
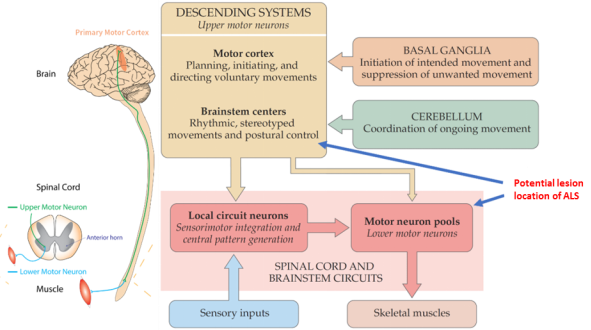
First, the motor cortex and brainstem in the brain transmit signals for active movement. The motor cortex is responsible for planning, initiating, and directing voluntary movements; the brainstem is responsible for rhythmic, stereotyped movements, such as breathing and postural control, which basically does not require the participation of active consciousness.
As supplementary areas of the brain, the basal ganglia is responsible for the initiation of intended movement and suppression of unwanted movement; the cerebellum is responsible for coordinating the functions of various departments during the whole process of movement, and the cerebellum is in charge of coordination of ongoing movement.
Signals from the motor cortex and brainstem are transmitted to the spinal cord or motor neuron pools. Some information goes directly to the motor ganglia, and some is transmitted to the motor neuron pools through the spinal cord.
Then, the motor neurons in the motor neuron pools begin to order the muscles to work. At the same time, the spinal cord is responsible for sensorimotor integration and central pattern generation through local circuit neurons, handling simple problems and reporting complex problems to the motor cortex or brain stem for instructions.
It should be noted that two new terms are introduced here, Upper Motor Neuron (UMN) and Lower Motor Neuron (LMN). The former refers to the cortex without direct communication with the muscles, namely the neurons in the brainstem and spinal cord (excluding neurons in the supplementary area such as the cerebellum and basal ganglia); the latter refers to the neurons connected to the motor nerves.
The potential lesion location of ASL occurs in the chain of command transmission formed by these two types of neurons. The muscles are not diseased, but atrophy as nerve activity decreases.
Classification of ALS
According to the classification of the motor nervous system, ALS can be divided into LMN-impaired and UMN-impaired types.
The key to distinguish the two is that the LMN-impaired type occurs directly in the neurons that communicate with the muscle from the beginning, so the symptoms are flaccid paralysis, hypomyotonia and hyporeflexia, convulsions and spasms, and a negative Babinski sign; the UMN-impaired type results in spastic paralysis, hypermyotonia and hyperreflexia, disuse-atrophy, and a positive Babinski sign.
Babinski sign:
It refers to a reflex elicited when the sole of the foot is stimulated with a blunt instrument. The reflex can take one of two forms. In healthy adults, the plantar reflex causes a downward response of the hallux (flexion). An upward response (extension) of the hallux is known as the Babinski response or Babinski sign, named after the neurologist Joseph Babinski. [2]
In addition, muscle atrophy of the LMN-impaired type occurs earlier and more rapidly than that of the UMD-impaired one, and the latter has a certain degree of muscle hyperfunction in the early stage.
The lesions of ALS are mainly located in motor neurons, but because its symptoms are similar to those of many cerebellar and muscle atrophy-related diseases, diagnosis is not easy. In particular, UMN-impaired ALS is often confused with primary lateral sclerosis (PLS), which is also a UMN-impaired disease. LMN-impaired ALS and progressive muscular atrophy (PMA) are also indistinguishable.
Some countries simply classify PLS and PMA as ALS. Due to unclear classification, many epidemiological statistical results vary.
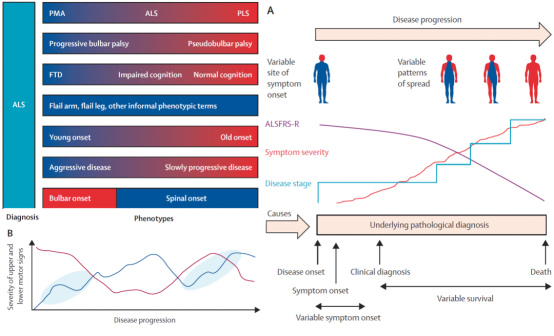
ALS is often accompanied by frontotemporal dementia. Patients without frontotemporal dementia, like Stephen Hawking, do not experience significant impairment in thinking ability.
There are also clear differences between early-onset and late-onset ALS in many cases. For example, a significant number of patients with early-onset disease survive beyond 10 years after diagnosis; and most late-onset patients have a very rapid course of disease, such as Stephen Hillenburg, the creator of SpongeBob SquarePants.
In addition, ALS and can be subdivided into bulbar-onset ALS and spinal-onset ALS. It is difficult to classify ALS because of its complex range of symptoms and pathological conditions, as well as the varying course of disease in different patients. Even among patients of the same age and with the same initial symptoms, the progression of disease and survival time vary widely. Therefore, it is very necessary and urgent to establish a clear ALS classification system.
Treatments
Currently, there are only two drugs on the market for ALS:
●Riluzole, which may increase survival by two to three months, has a relatively good effect on bulbar-onset ALS and performs well in the early stages of the disease. The mechanism has not yet been fully elucidated, and it is currently attributed to the inhibitory effect of presynaptic glutamate, but the inhibition of excitatory neurotransmitters can cause mental and motor weakness in patients. The treatment of ALS with Riluzole requires many drugs to control its complications.
●Edaravone, a drug from Japan, can reduce the incapacitation of bodily functions in ALS, but is only effective in a small percentage of early-onset ALS. The mechanism may be to protect neurons by inhibiting excessive oxidation of cells. Edaravone also has serious side effects and costs as much as $140,000 a year.
Other drugs can only relieve the pain of patients, and their effects mainly include reducing neuralgia and fatigue, relaxing muscles, anti-spasticity, suppressing external symptoms such as drooling and phlegm. Besides, there are anti-depressants, antianxiety drugs and other psychotropic drugs, as well as some drugs that are helpful for cognitive function.
There are also assistant treatments that help relieve ALS symptoms:
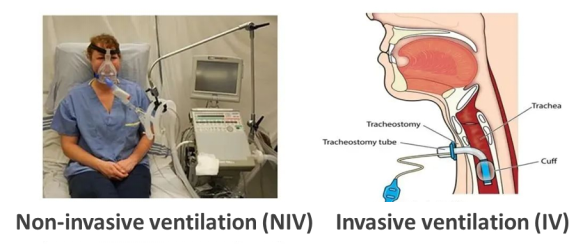
●Non-invasive ventilation (NIV) is an assistant method to prevent suffocation during sleep in patients who have lost the ability to spontaneous respiration in an advanced disease stage. The principle is that the patient wears a mask that covers the nose and mouth, and uses a pump to press air into the lungs at a set frequency to assist breathing. Studies have shown that concomitant use of NIV has a better effect on lifespan than Riluzole alone, and the results of quality of life assessment are also better. However, patients with severe cognitive impairment and severe bulbar impairment cannot receive this treatment.
●Invasive ventilation (IV) refers to the administration of ventilatory support using an invasive artificial airway (endotracheal tube or tracheostomy tube), which is suitable for patients with weaker respiratory function who do not respond to NIV. However, the quality of life of patients using this treatment is greatly reduced, and the cost is extremely high. The acceptance of IV varies from country to country. In Japan, 30% of patients are willing to receive it, while only 5% of those in Europe and the United States are willing to live a few more months in this way.
RDDC Aids the Research of Rare Diseases
The Rare Diseases Data Center (RDDC) was developed by Research Institute of Tsinghua, Pearl River Delta (RITPRD) and assisted by Cyagen Bioscience.
RDDC has collected and sorted out data and information about genes, mutations, phenotypes and animal models of various types of rare diseases from international public biological resources, aiming to present them in a visual way. At present, it has collected over 20,000 gene information and 14,000 disease information!
Users can not only better understand the information of corresponding rare diseases, but also can take advantage of various AI tools to predict pathogenicity and RNA splicer, so as to help speed up the diagnosis and development of treatment research of rare diseases.

Click the photo above for more information!
*Statement: RDDC data and tools are only for scientific research and are only for reference, and cannot be used as the final conclusion of medical diagnosis and evaluation.
Reference:
[1] Dale Purves et al. Neuroscience.Sinauer Associates.2018.9781605353807,1605353809
[3] Al-Chalabi A , Hardiman O ,Kiernan M C , et al. Amyotrophic lateral sclerosis: moving towards a newclassification system[J]. Lancet Neurology, 2016, 15(11):1182-1194.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



