

뒤셴근이영양증(Duchenne muscular dystrophy, DMD)은 진행성 근육 변성을 특징으로 하는 유전 질환입니다. 이번 주에 Cyagen 칼럼 <Gene of the Week>에서 우리는 근이영양증단백질(DMD)의 단백질 코딩 유전자(뒤셴근이영양증의 원인 유전자)에 대한 배경 정보와 연구 견해를 검토했습니다.
DMD 유전자는 디스트로핀 단백질의 코딩을 담당합니다. DMD 단백질은 근육막의 디스트로핀 관련 단백질 복합체(DAPC)와 액틴 세포골격 사이의 중요한 연결을 제공하기 때문에 근육막의 안정성에 필요합니다. 이 단백질은 완충 연결 역할을 하고 근육 세포를 주변 구조에 고정시킵니다. DMD 단백질이 부족하면 DAPC가 분해되고 액틴과 세포외 기질 사이의 상호 작용이 손실되어 근육 섬유가 기계적 손상에 취약하게 됩니다[1]. DMD 유전자는 인간 염색체 X에 있습니다. 유전자의 길이는 약 2.2Mb이고 89개의 엑손과 3685개의 아미노산이 있습니다. 이 유전자의 돌연변이는 뒤셴근이영양증(Duchenne muscular dystrophy,DMD )과 관련이 있습니다.
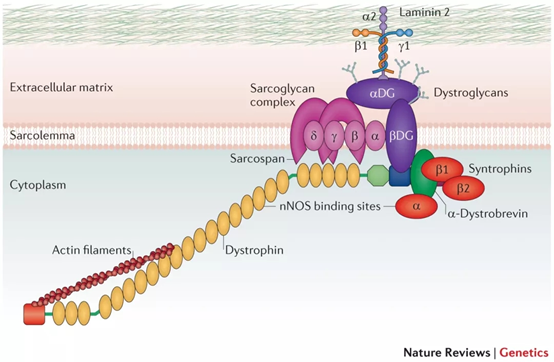
그림 1. 디스트로핀은 내부 세포골격과 세포외 기질 사이의 중요한 연결 고리 역할을 합니다[1].
|
Species |
Human |
Mouse |
Rat |
|
Chromosome |
X |
X |
X |
|
Full Length (bp) |
2220242 |
2390413 |
2231896 |
|
mRNA (nt) |
13,854 |
13852 |
13819 |
|
Number of Exons |
89 |
85 |
81 |
|
Number of Amino Acids |
3685 |
3678 |
3699 |
|
Gene Family |
BMD, CMD3B, DXS164, |
||
|
Cyagen Mouse Models |
|||
|
Status |
Custom |
Catalog Models |
Live Mice |
|
Knockout (KO) |
√ |
|
|
|
Conditional Knockout (cKO) |
√ |
|
|
뒤셴근이영양증(Duchenne muscle dystrophy, DMD)은 심각한 X염색체 열성 유전질환으로 DMD 유전자의 돌연변이에 의해 근육에서 DMD 단백질 생성이 차단되어 근위축을 유발하여 많은 합병증을 유발합니다. 베커 근이영양증(BMD)은 DMD와 마찬가지로 DMD 유전자의 돌연변이에 의해 발생하지만 돌연변이 부위가 다르기 때문에 일부 기능적 DMD 단백질이 여전히 생성될 수 있으므로 BMD 환자의 증상은 DMD 환자에 비해 가볍습니다.
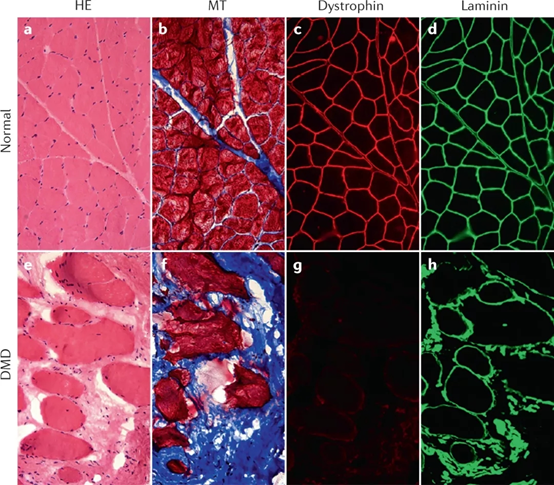
그림 2. 건강한 골격근의 단면 염색(a-d) 및 DMD 환자의 골격근 단면 염색(e-h) [2]
DMD는 주로 남성에게 발생하며, 평균적으로 전 세계 남자 아기 5,000명 중 1명이 이 질병을 앓고 있습니다. 환자는 2~3세 경에 거동이 불편한 등의 초기 증상을 나타내며, 대부분의 환자는 10~12세 경에 휠체어에 의존하기 시작하여 20세 경에 호흡 보조가 필요합니다. 최선의 치료 하에 대부분의 DMD 환자는 20세에서 40세 사이에 심부전 또는 호흡 부전으로 사망합니다.[2]
DMD의 가장 널리 사용되는 동물 모델은 30년 전에 처음 발견된 디스트로핀 결핍 mdx 마우스로 DMD 환자와 마찬가지로 디스트로핀 유전자 자체에 돌연변이가 있어 근육이 완전 기능 디스트로핀의 발현이 감소합니다. 마우스에서 Dmd mdx 돌연변이는 엑손 23에 종결코돈이 있어 절단된 단백질을 생성할 수 있고 DMD의 근병리학적 특성을 갖지만, 그 근병리학적 상태는 인간보다 온화하고 마우스 계통 유전적 배경에 따라 변화가 발생됩니다.
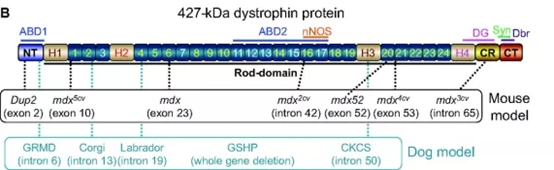
그림 3. 마우스 모델에서 돌연변이의 위치[3]
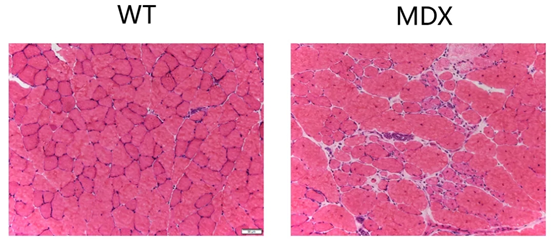
그림 4. mdx 마우스 모델의 근육 단면[3]
현재, DMD의 전반적인 치료 방향은 주로 기능적 디스트로핀 발현의 회복과 DMD의 발병 기전을 표적화하는 방법의 두 가지 측면으로 나뉩니다. 유전자 추가, 엑손 건너뛰기, 정지 코돈 판독 및 게놈 편집과 같은 치료는 기능성 DMD 단백질의 일부의 발현을 회복시켜 환자의 근섬유 손상 및 위축을 감소시킬 수 있습니다. 유전자 추가 요법은 주로 AAV와 같은 바이러스 벡터를 사용하여 기능적 DMD의 cDNA 사본을 영향을 받은 조직에 전달하여 조직이 DMD 단백질을 재발현하도록 합니다. 엑손 건너뛰기는 변형된 RNA 또는 DNA의 작은 단편을 사용하고 안티센스 올리고뉴클레오티드라고 합니다. 이들은 pre-mRNA 디스트로핀 전사체의 표적 엑손에 있는 특정 서열에 결합합니다. 이 조합은 표적 엑손을 건너뛰게 하여 판독 프레임을 복원하고 비기능성 디스트로핀 아닌 기능성 디스트로핀의 일부를 생성합니다.
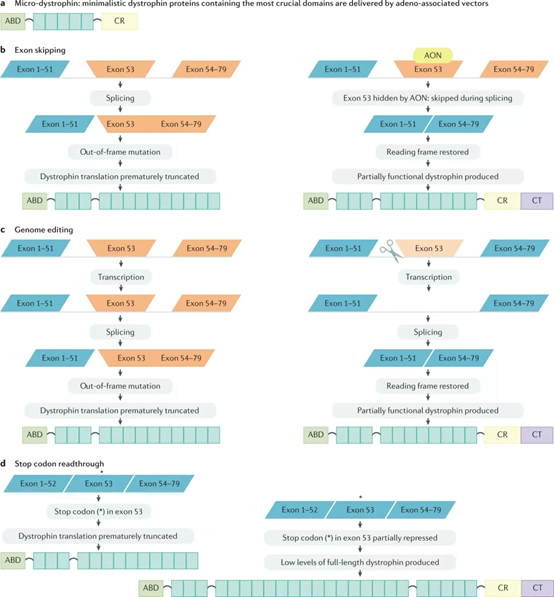
그림 5. DMD의 유전자 치료[2]
DMD 단백질의 손실은 염증, 칼슘 항상성의 손실, 기능적 허혈 및 손상된 근육 재생을 비롯한 다양한 병리학적 경로를 유발할 수 있습니다. 기능적 DMD 발현 감소를 교정하기 위한 치료법 외에도 현재 임상 시험에서는 DMD 단백질 발현 감소로 인한 속발성 병리의 화합물로 근육 기능과 질을 개선하는 것을 표적으로 연구하고 있습니다. 주로, 항염증 화합물, 항산화제, 혈관 확장제를 포함합니다.
DMD 돌연변이는 Duchenne 근이영양증의 주요 원인입니다. 주요 원인은 기능적 DMD 단백질의 결핍이지만 발병 기전은 매우 이질적입니다. 여러 환자들의 체내 DMD 유전자에 여러가지 돌연변이가 있었습니다. 현재의 유전자 요법은 초기 결과를 달성했지만 몇 가지 어려움과 과제가 해결되지 않은 채로 남아 있습니다. 첫째로, 이러한 마이크로디스트로핀의 인체 내 기능이 불분명하고, 엑손 조합이 다른 DMD 단백질의 생물학적 기능이 다릅니다. 예를 들어, BMD 환자 체내에서 생성된 DMD 단백질은 부분적인 기능적 활성을 가지고 있습니다.또한 일부 돌연변이 개체에 의해 생성된 DMD 단백질은 더 많은 엑손을 포함할 수 있으나 단백질 활성이 완전히 결여되어 근육 위축의 증상이 오히려 악화됩니다. 둘째로, 형질전환의 외부 도움을 통한 DMD 단백질의 발현 역시 한계가 있으며, 형질전환 유전자 발현의 수명이 제한적이며, 바이러스 벡터의 대량 생산에 많은 시간과 비용이 소요됩니다. 엑손 건너뛰기 방법은 pre-mRNA 전사체를 표적으로 하고 안티센스 올리고뉴클레오티드와 RNA 전사체는 쉽게 분해되기 때문에 안티센스 올리고뉴클레오티드를 반복적으로 사용해야 합니다.
모델 동물을 기반으로 한 국제화된 혁신적인 CRO 플랫폼으로서 Cyagen은 희귀질환이 모든 인류가 직면하고 있는 일반적인 공중 보건 문제라는 것을 알고 있으며 자신의 전문 분야 기술을 통해 희귀질환 유전자 치료 연구에 도움이 되기를 희망합니다. 따라서 Cyagen은 ‘희귀질환 유전자 디코딩 프로그램’을 본격적으로 시작해 희귀질환을 유발하는 유전자 데이터를 보완하고, 강력한 데이터 분석 플랫폼 및 원스톱 마우스 모델 서비스 플랫폼과 결합하여 전 세계 고객들과 희귀질환 모델을 공동 개발해 희귀질환 치료제 개발을 지원하는 데 주력하고 있습니다.
희귀질환 관련 연구를 하고 계시거나 관심있는 분은 연락 주시기 바랍니다. 우리는 귀하와 함께 희귀질환 마우스/랫드 모델을 공동 개발할 것이며 관련 동물 모델은 초저가 또는 증여 방식으로 제공됩니다.
Cyagen의 AI Knock-Out Mouse Model eBank에서는 16,000 strains 이상의 KO/cKO 마우스 모델을 보유하고 있으며, 연구자들의 연구 진행에 도움이 되도록 최적의 마우스 모델을 제공한다.
>> Search Your Gene of Interest
>> Live KO/cKO Mice Shipped in 2 Weeks



영업일 기준 1-2일 내에 답변해 드리겠습니다.



