

눈은 영혼을 보여주는 창이라고 하지만, 세상에는 선천적으로 앞을 '볼’ 수 없는 시각 장애인이 많습니다. 세계보건기구의 공개 데이터에 따르면, 전 세계적으로 시각 장애 또는 실명 상태에 있는 인구는 최소 22억 명에 달합니다. 특히, 중국의 경우 전 세계 시각 장애인 인구의 18~20%를 차지할 정도로 세계에서 시각장애인 수가 가장 많은 국가입니다. 유전성 안구 질환은 소아 실명 및 시각 장애 사례의 약 3분의 1을 차지하며, 실명 및 중증 시각 장애 사례의 80%를 차지합니다. 선천성(유전성) 안구 질환은 현재 소아 및 청소년 실명의 가장 흔한 원인으로, 후대의 시력에 가장 큰 위협이 되는 질환임이 분명합니다.
유전성 망막 질환(Inherited Retinal Disease, IRD)은 망막에 영향을 미치는 이종 유전 질환군을 일컫습니다. 이는 광수용체(photoreceptor), 망막 색소 상피(retinal pigment epithelium) 또는 맥락막(choroid)의 기능 장애 및 퇴화를 야기해 시각 장애를 초래합니다. IRD와 관련된 병원성 유전자는 300가지가 넘습니다. 그러나 현재 수행되는 연구는 주로 망막 색소 변성증(Retinitis Pigmentosa), 레버 선천성 흑암시(Leber Congenital Amaurosis), 스타가르트병(Stargardt Disease) 등 일반적인 광수용체 IRD와 황반변성(macular degeneration) 질환에 초점을 맞추고 있습니다.
많은 희귀 IRD는 여전히 치료가 불가능하며, 치료를 받지 않으면 환자는 중증 시각 장애와 실명으로 고통받게 됩니다. 대부분의 희귀 IRD는 단일 유전자적인 특성을 가지고 있고 망막은 아데노 부속 바이러스(Adeno-Associated Virus, AAV)와 같은 유전자 치료 벡터의 효율적인 표적 부위이기 때문에, 안과 질환에 대해 사용할 수 있는 표준화된 약리학적 유효성 평가 방법이 점점 증가하고 있습니다. 이에 따라, 유전자 치료는 IRD와 같은 희귀 안구 질환을 앓고 있는 환자들에게 새로운 희망으로 빠르게 부상하고 있습니다.
이러한 맥락에서, 당사는 몇 가지 희귀 IRD에 대한 유전자 치료 접근법의 개발 사항을 소개하고, 전임상 약물 유효성 평가를 위한 신뢰할 수 있는 전임상 동물 질환 모델을 권장합니다.
뇌회형 맥락막 및 망막 위축(Gyrate Atrophy Of Choroid & Retina, GACR)은 아미노산 대사 장애로 분류되는 희귀 상염색체 열성 유전 질환입니다. 이는 유전자를 코딩하는 미토콘드리아 효소인 오르니틴 아미노 전달 효소(Ornithine Aminotransferase, OAT)의 병원성 돌연변이로 인한 맥락막의 퇴행 및 위축이 특징입니다. OAT는 주로 간에서 발현되며, GACR을 가진 사람은 혈액과 기타 체액에서 오르니틴 수치가 높게 나타납니다. 환자는 진행성 시야 협착을 경험하여 결국 실명에 이르게 됩니다. 초기 증상으로는 일반적으로 야맹증과 말초 맥락막 및 망막 퇴행으로 인한 시야 수축 등이 있습니다. 말기에는 심각한 중심 시력 손실이 발생할 수 있으며 황반이 영향을 받는 경우 완전 실명이 발생할 수 있습니다. 현재 치료법은 없지만, 일부에서는 오르니틴 수치가 높으면 망막의 섬세한 구조가 부정적인 영향을 받을 수 있으며, 오르니틴 수치를 낮추기 위한 조기 중재가 잠재적으로 질환 진행을 예방하거나 지연시킬 수 있다고 제안합니다[1-2].
OAT가 결핍된 마우스는 신생아 저오르니틴혈증(neonatal hypoornithinemia)과 치사율을 보이지만, 아르기닌을 단기간 보충하면 회복될 수 있습니다. 이러한 마우스에서는 이유(weaning) 후, 인간 GACR 환자와 유사한 고오르니틴혈증이 발생합니다. 이러한 마우스에서는망막 퇴행이 서서히 진행되며, 이는 망막 전도(electroretinogram, ERG) 진폭의 점진적인 감소로 나타납니다. 시간이 경과함에 따라, 망막 색소 상피(retinal pigment epithelium)에 이상이 나타나고 광수용체(photoreceptor)의 외부 분획이 짧아지고 구조가 해체되며 광수용체(photoreceptor) 세포가 점진적으로 소실됩니다. OAT 결핍 마우스는 인간 GACR과 유사한 질환 표현형을 나타내기 때문에, 우수한 전임상 동물 모델입니다[3-4].
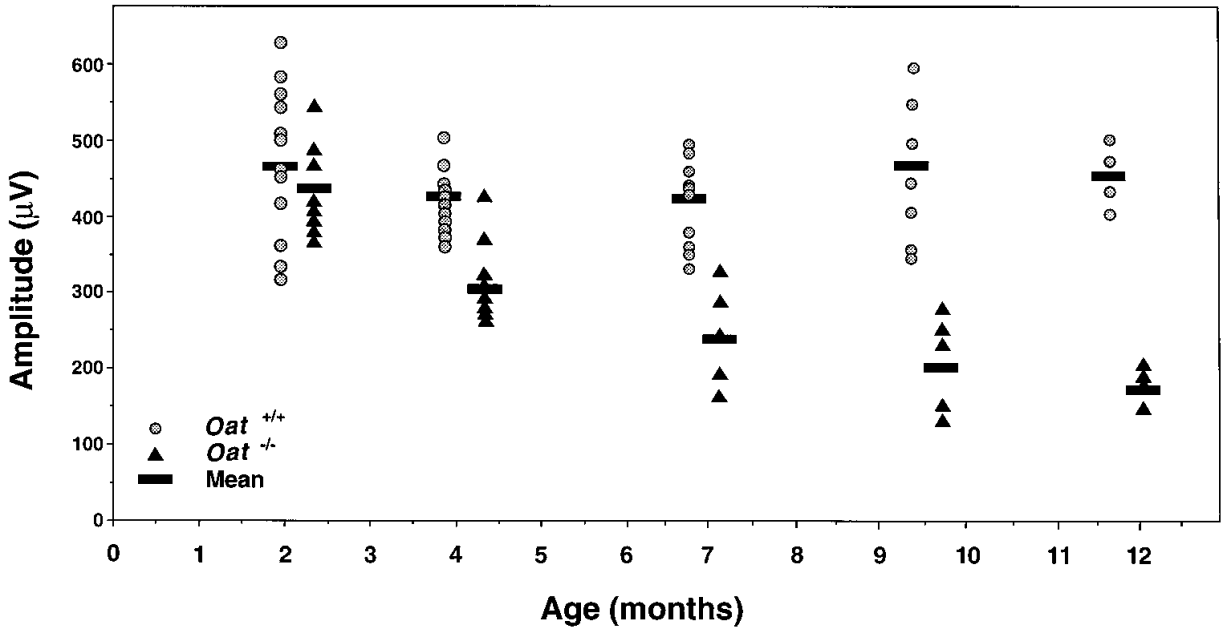
그림 1. OAT 결핍 마우스(Oat-/-)의 망막 전도(Electroretinogram, ERG) 진폭이 현저히 감소했습니다[3]
유전자 치료는 다양한 단일 유전자 질환에 대한 효과적인 해결책으로 부상했습니다. 최근 연구에서는, 치료를 위해 AAV8 벡터를 사용하여 간 특이적 프로모터를 전달하여 OAT 유전자를 조절했습니다. 이 유전자 치료 벡터를 투여한 결과, OAT 결핍 마우스의 혈액과 안구에서 오르니틴 수치가 감소하고 망막 구조가 부분적으로 회복되었으며 망막 전도(electroretinogram)가 개선된 것으로 나타났습니다. 이러한 효과는 1년 이상 지속되었습니다.[5]. 이 연구에 따르면, AAV8-OAT 유전자 치료가 고오르니틴혈증을 효과적으로 교정하고 망막의 기능과 구조를 모두 개선하는 것으로 나타났습니다. 이는 GACR에 대한 간 표적 AAV 매개 유전자 치료의 효과를 개념적으로 입증합니다.
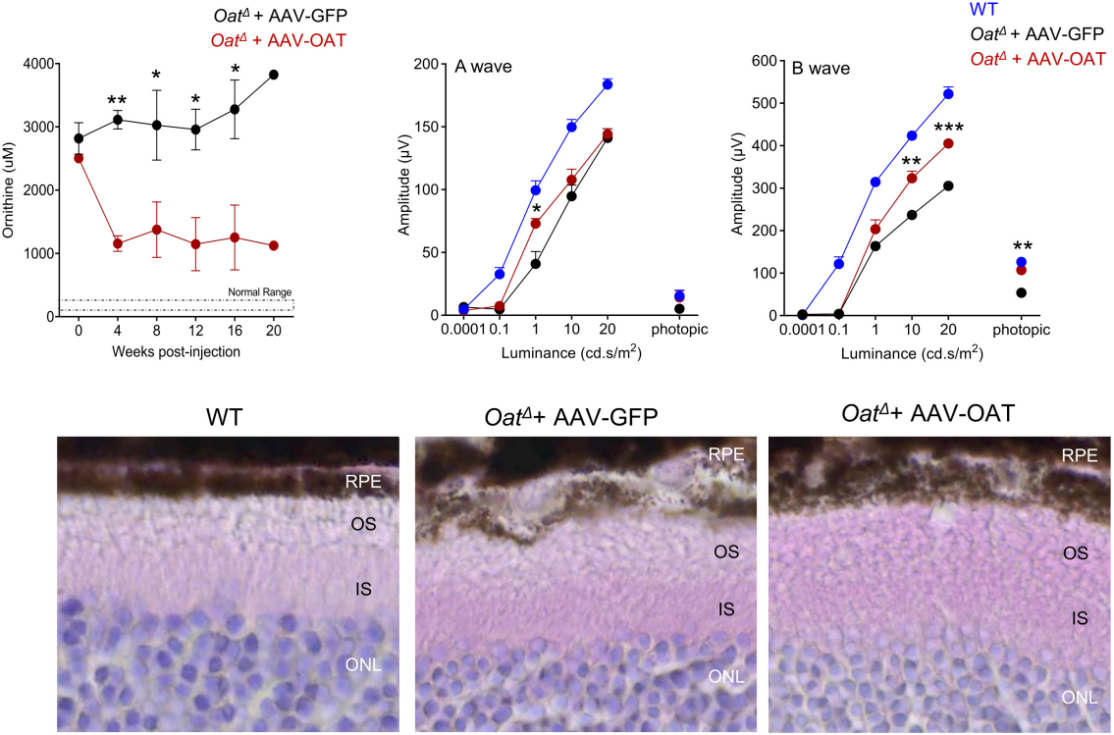
그림 2. AAV 매개 간 특이적 OAT 유전자 치료는 OAT 결핍 마우스(OatΔ)에서 질환 표현형을 개선합니다[5]
어셔 증후군(Usher Syndrome)은 유전성 난청-망막 색소 변성증 증후군(Genetic Deafness-Retinitis Pigmentosa Syndrome)으로도 알려져 있으며, 상염색체 열성 유전성 유전 질환으로, 망막 색소 변성증(retinitis pigmentosa, RP)과 다양한 정도의 청력 손실을 특징으로 합니다. 이 유전적 이종 질환군 중 제3형 어셔 증후군(Usher Syndrome Type III, USH3)은 가장 희귀한 유형입니다. USH3는 청소년기의 진행성 감각신경성 난청(progressive sensorineural hearing loss)과 망막 색소 변성증(retinitis pigmentosa)을 특징으로 합니다. 클라린-1(clarin-1, CLRN1) 유전자는 USH3의 발병과 관련된 유일한 유전자로 알려져 있습니다.
제3형 어셔 증후군(Usher Syndrome Type III) 환자는 일반적으로 출생 시 청력이 정상이지만, 대개 유년기 후반이나 청소년기에 청력 손실을 경험하기 시작하여 시간이 경과함에 따라 악화됩니다. 청각 표현형과 마찬가지로, 안구 표현형도 진행성으로 나타납니다. 시각 증상은 청소년기 무렵에 나타나는 경우가 많으며, 많은 환자가 처음에 야맹증을 경험한 후 시야가 점차 감소하여 터널 시야(tunnel vision)를 경험합니다. 이후, 중심 시력과 색 지각 능력을 상실하여 결국 완전 실명으로 진행됩니다[6].
마우스에서 Clrn1 유전자의 녹아웃(KO)은 청각 장애와 망막 퇴행을 초래합니다. 특정 안구 표현형으로는 시각 장애와 시야 협착을 야기하는 망막의 광수용체(photoreceptor) 세포 수 감소 등이 있습니다. 이러한 증상은 일반적으로 마우스의 출생 후 초기 단계에 나타나며 시간이 경과함에 따라 악화됩니다. 또한, Clrn1-KO 마우스는 생후 2일째부터 유모 세포(hair cell) 결함이 나타나고 생후 21~25일경에 청각 장애가 발생합니다.
연구자들은 UTR 영역을 포함하는 Clrn1 유전자를 도입하여 Clrn1-KO 마우스에서 유모 다발(hair bundle) 구조의 악화와 관련된 후기 발병 진행성 난청을 유도할 수 있었습니다. Clrn1-KO 마우스는 시각 및 청각 장애 외에도, 균형 장애 및 운동 협응 장애와 같은 다른 계통에서 표현형 변화도 보일 수 있으며[7-10], 이는 인간 USH3 표현형과 유사합니다.
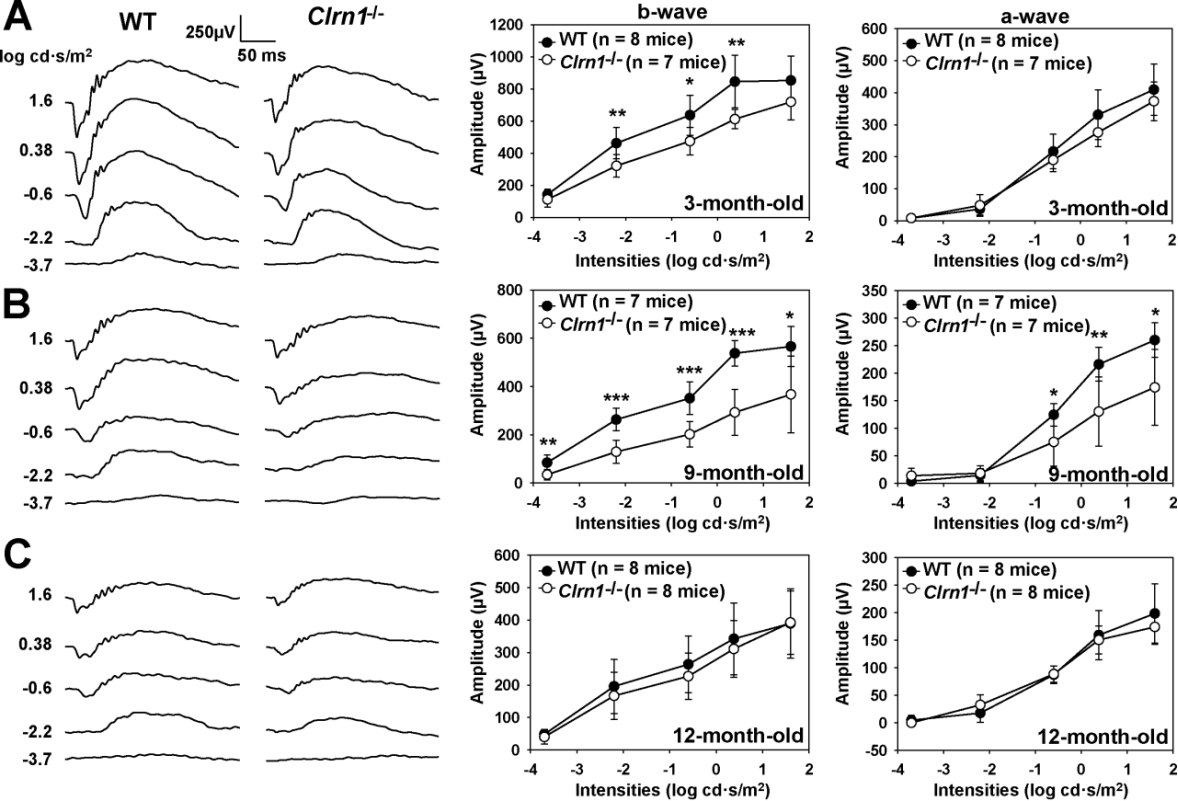
그림 3. Clrn1-/- 녹아웃 마우스(Clrn1-/-)의 결함성 망막 전도(Electroretinogram, ERG) 진폭(Clrn1-/-) [8]
이와 유사하게, 단일 유전자 질환으로서, USH3를 표적으로 하는 유전자 치료법이 개발되고 있습니다. USH3에 대한 AAV 기반 유전자 치료에 관한 여러 연구가 발표되었습니다[9-11]. Dinculescu et al.이 Clrn1-KO 마우스를 사용하여 생체 내에서 AAV-CLRN1 유전자 치료의 가능성을 연구했습니다. 이들의 연구에 따르면, 모든 주요 계통의 망막 세포가 공통 구성 닭 β-액틴 프로모터에 의해 유도되는 AAV-CLRN1을 발현하는 것으로 나타났습니다. 외인성 CLRN1은 주로 내인성 단백질의 발현 패턴과 유사하게 내부 분획 영역과 외부 결합층에 국한되어 있으며, 이는 향후 USH3 유전자 치료 연구를 설계하는 데 매우 중요합니다.
그러나 최대 역가의 바이러스를 망막하로 전달한 결과 망막 기능이 크게 손실되었으며, 이는 광수용체(photoreceptor) 세포에서 CLRN1 발현에 중요한 임계치가 존재함을 시사합니다. 이러한 결과는 안전한 USH3 치료 접근법을 개발하기 위해 올바른 AAV 벡터 용량, 프로모터, 전달 방법을 신중하게 선택해야 함을 시사합니다[11].
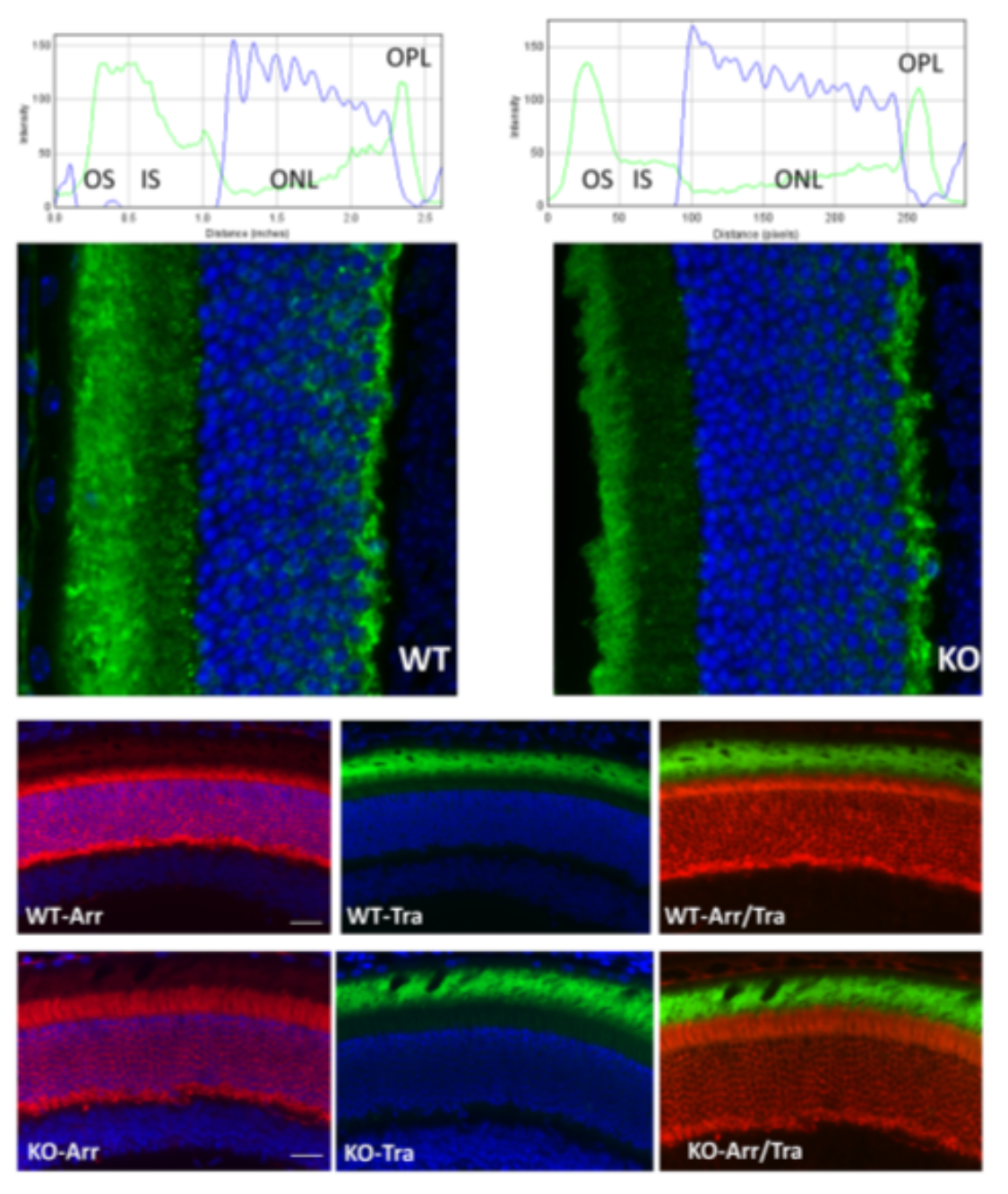
그림 4. Clrn1-녹아웃 및 야생형(Wild-Type) 마우스의 망막 단백질 발현에 대한 AAV-CLRN1 유전자 치료의 효과[11]
범맥락막 위축(Choroideremia, CHM)은 희귀한 X 연관 열성 유전 질환으로, 전 세계적으로 약 5만 명 중 1명에서 10만 명 중 1명의 발병률을 보입니다. 이 질환의 주요 증상으로는 중심 시력의 점진적인 저하, 시야 협착, 색각 이상 등이 있습니다. 범맥락막 위축(Choroideremia)은 X 염색체에 위치한 CHM 유전자의 돌연변이로 인해 발생하며, 여성보다 남성에서 흔하게 발생합니다. CHM 유전자는 망막 색소 상피 세포에서 중요한 역할을 담당하는 막 골격 단백질인 랍 에스코트 단백질-1(Rab escort protein-1, REP1)을 부호화합니다. REP1의 손실 또는 기능 장애는 망막 색소 상피 세포의 퇴화와 망막 위축으로 이어져 범맥락막 위축(choroideremia)을 유발합니다. 현재로서는 범맥락막 위축(choroideremia)을 치료할 수 있는 방법이 없으며, 치료는 증상 관리에 초점을 맞추고 있습니다[12]. 동물 실험에서, Chm/REP1의 전신 또는 망막 특이적 녹아웃(KO) 마우스는 광수용체(photoreceptor)의 진행성 퇴행, 결함성 랍 프레닐화(defective Rab prenylation), 망막 색소 상피 세포 퇴행, 망막 위축, 망막 혈관 이상, 시각 장애 등 안구와 관련된 표현형 이상을 보였습니다[13-14].
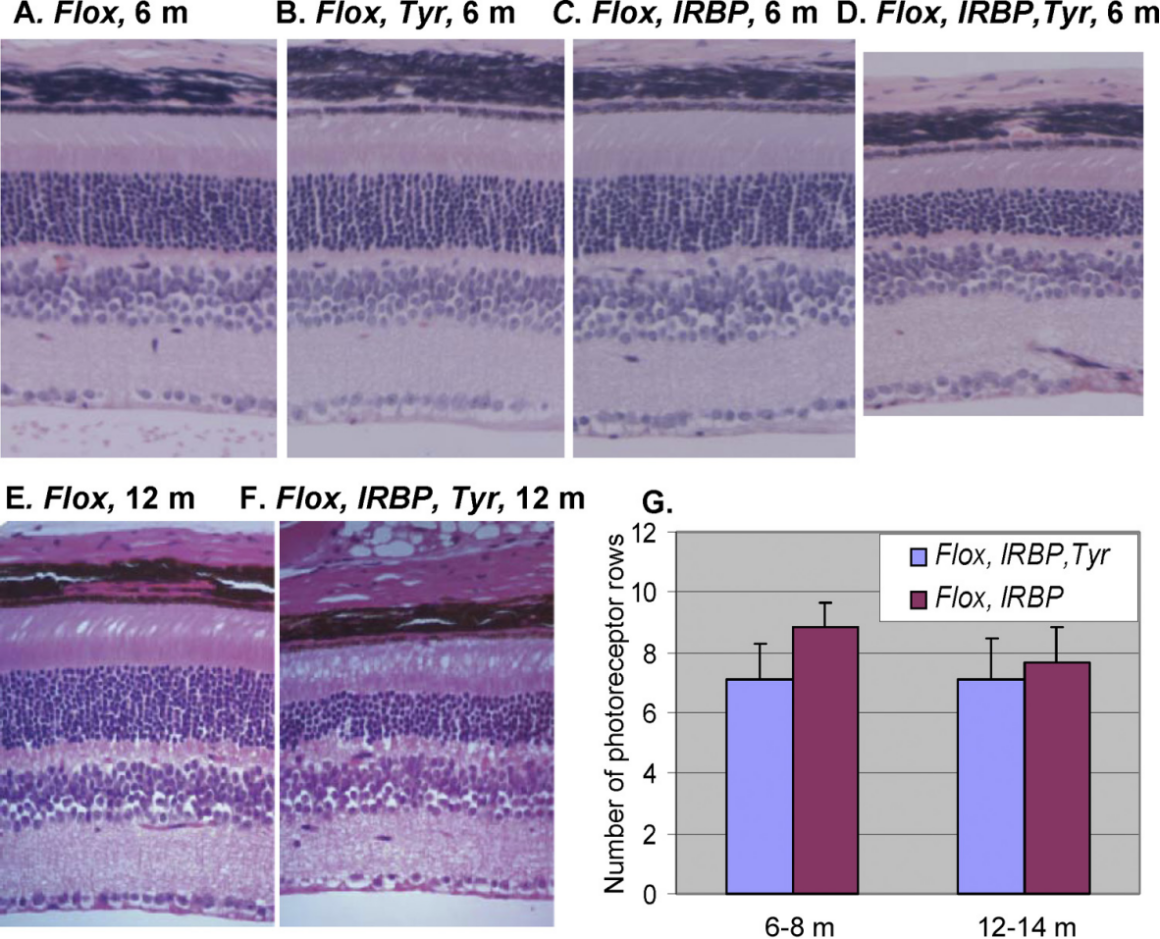
그림 5. 망막 특이적 Chm/Rep1의 녹아웃은 광수용체(photoreceptor)의 중증 퇴행을 초래합니다[14]
현재, CHM을 발현하는 아데노 연관 바이러스(adeno-associated virus, AAV) 벡터의 망막하 전달을 매개로 한 유전자 치료를 범맥락막 위축(choroideremia) 치료로 평가하는 임상시험이 8건 이상 진행 중입니다. 대표적인 예로는, Biogen의 BIIB111/AAV2-REP1과 4D Molecular Therapeutics의 4D-110이 있습니다[15]. BIIB111/AAV2-REP1은 3상 임상시험인 STAR 임상시험에서 1차 평가변수 또는 주요 2차 평가변수를 충족하지 못했지만, STAR 임상시험 이전의 임상 및 전임상 데이터는 향후 CHM 치료를 위한 유전자 치료 접근법에 대한 가치 있는 참고 데이터를 제공합니다. 이 임상시험을 통해 얻은 인사이트는 범맥락막 위축(choroideremia)을 비롯한 유전성 망막 질환에 대한 혁신적인 치료법을 개발하는 데 기여합니다. 이러한 약물에 대한 전임상 연구에서, Chm/Rep1 유전자 녹아웃(KO) 마우스는 여전히 전임상 약물 유효성 평가에 필수적인 모델로 활용되고 있습니다. 한 연구에서, AAV2/2-CBA-REP1 유전자 치료는 Chm/Rep1 유전자 KO 마우스의 망막에서 암순응 ERG의 a파 및 b파 진폭에 용량 의존적인 영향을 미쳤습니다. 고용량의 AAV2/2-CBA-REP1 치료는 Chm/Rep1 유전자 KO 마우스에서 암순응 망막 기능을 개선하는 것으로 나타났습니다[16].
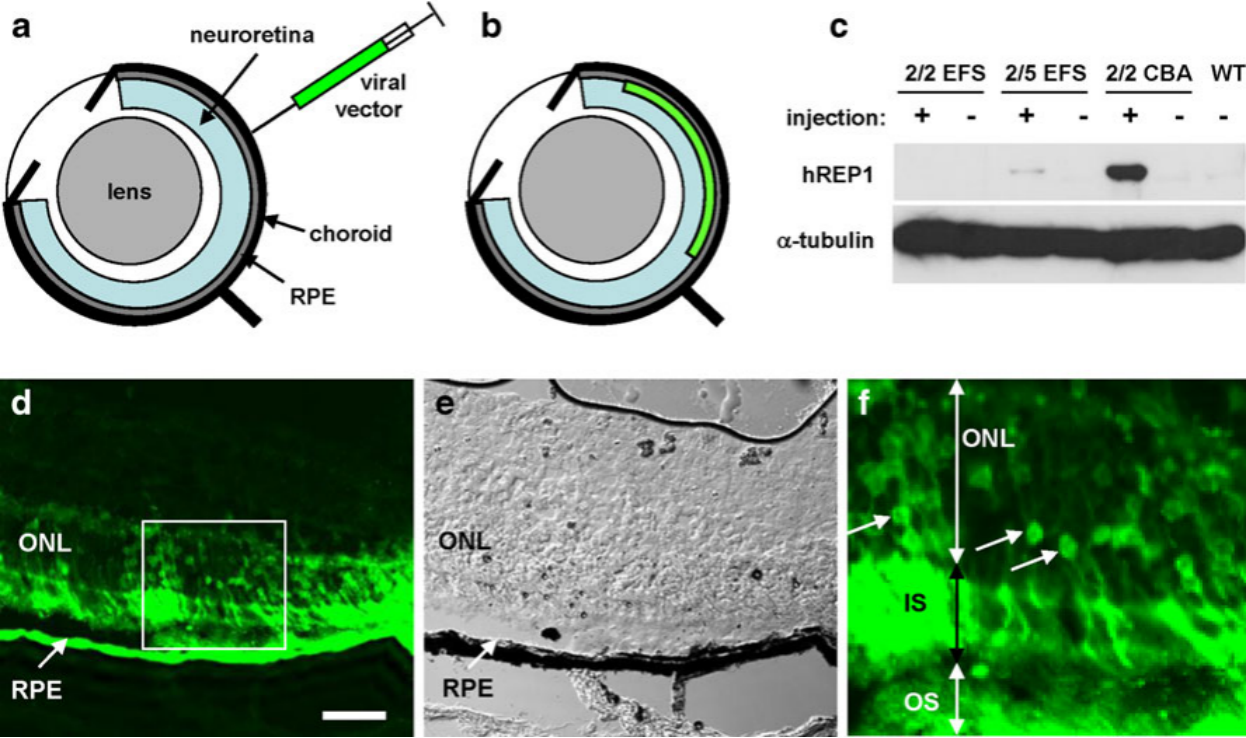
그림 6. Chm/Rep1 유전자 녹아웃 마우스를 활용한 AAV2/2-CBA-REP1의 유효성 평가 실험[16]
유전자 변형 마우스 모델은 희귀 질환 기전 연구와 전임상 약물 개발 및 평가에서 중요한 역할을 합니다. Cyagen은 희귀 질환에 대한 다양한 유전자 녹아웃(KO) 및 조건부(예: 조직 특이적) KO(cKO/floxed) 모델 등 독자적으로 개발한 OAT, CLRN1, CHM 등의 수천 가지 게놈 변형 마우스 계통 품종(strain)을 제공하고 있습니다. 또한, 당사의 맞춤형 동물 모델(CAM) 개발 및 CRO 서비스는 연구 요구사항에 맞게 맞춤화할 수 있으며, 포괄적이고 신속한 턴어라운드를 제공하는 CAM/CRO 솔루션으로 희귀 질환 연구를 가속화할 수 있습니다.
|
질환 |
표적 유전자 |
유형 |
|
뇌회형 맥락막 및 망막 위축(Gyrate Atrophy Of Choroid & Retina, GACR) |
Oat |
KO, CKO |
|
제3형 어셔 증후군(Usher Syndrome Type III, USH3) |
Clrn1 |
KO |
|
범맥락막 위축(Choroideremia, CHM) |
Chm |
KO, CKO |
Cyagen은 종합 임상시험수탁기관(contract research organization, CRO) 서비스 제공업체로서, 안과 질환을 유전자 치료의 돌파구로 인식하고 전임상 연구를 지원하기 위해 안과 유전자 치료 플랫폼을 설립했습니다. 당사의 숙련된 전문가 팀은 소동물용 최첨단 안과 장비 제품군을 활용하여 표준화된 평가 서비스를 수행할 수 있습니다. 당사의 안과 기술 제품에는 Micron IV 소형 동물 망막 현미경 영상 촬영 시스템, 전체 시야 망막 전도(full-field electroretinogram, ffERG), 영상 유도 빛 간섭 단층 촬영(optical coherence tomography, OCT) 시스템, 마우스용 휴대용 안압계(ophthalmotonometer) 등이 있습니다. 당사는 당뇨망막병증(당뇨망막병증), 망막모세포종(retinoblastoma), 황반변성(macular degeneration), 소아 미숙아망막증(retinopathy of prematurity, ROP), 맥락막신생혈관(choroidal neovascularization), 망막 색소 변성증(retinitis pigmentosa) 등 안구 관련 질환의 설치류 모델에 대한 검출 서비스를 제공할 수 있습니다. Cyagen은 16년에 이르는 유전자 편집 모델 구축 경험을 바탕으로, 안과 유전자 치료를 위한 다양한 표준화된 전임상 연구 솔루션을 제공할 수 있습니다.
Cyagen은 유전자 치료제 개발을 가속화하기 위해 차세대 인간화 마우스 모델 개발 프로그램을 출시했습니다. 여기에는 당사의 새로운 전체 게놈 DNA 유전자 치료를 위한 인간화 게놈 동원체(Humanized Genomic Ortholog)(HUGO-GTTM) 마우스가 활용되며, 병원성 유전자에 대한 전체 게놈 커버리지를 제공하여 ASO/CRISPR/siRNA를 포함한 유전자 치료의 효율적인 약물 스크리닝을 지원합니다. Cyagen은 질환-유전자-동물 모델-약물 임상 정보에 접근할 수 있는 종합적인 생물학적 데이터 플랫폼인 희귀 질환 데이터 센터(RDDC)를 기반으로, 대규모 기능에서 완전한 게놈 대체 및 인간화 기능으로 전환하기 위해 터보녹아웃(TurboKnockout) 기술과 BAC 융합 기술을 활용했습니다. 그 결과 희귀 선천성 질환 연구와 유전자 치료 약물 개발에 더 적합한 전체 게놈 인간화 모델을 개발할 수 있었습니다.
Cyagen은 안과 질환 분야에서 야생형 전체 게놈 인간화 마우스 hRHO를 포함한 전체 게놈 인간화 마우스와 이를 기반으로 구축된 hRHO-P23H 점 돌연변이 모델 및 hCEP290 등의 기타 안과 모델과 같은 관련 안과 질환 모델을 독자적으로 개발했습니다.
참고문헌:
[1]Pauleikhoff L, Weisschuh N, Lentzsch A, Spital G, Krohne TU, Agostini H, Lange CAK. Clinical characteristics of gyrate atrophy compared with a gyrate atrophy-like retinal phenotype. Eur J Ophthalmol. 2023 May 23:11206721231178147.
[2]Elnahry AG, Tripathy K. Gyrate Atrophy of the Choroid and Retina. 2023 Apr 10. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan.
[3]Wang T, Milam AH, Steel G, Valle D. A mouse model of gyrate atrophy of the choroid and retina. Early retinal pigment epithelium damage and progressive retinal degeneration. J Clin Invest. 1996 Jun 15;97(12):2753-62.
[4]Wang T, Lawler AM, Steel G, Sipila I, Milam AH, Valle D. Mice lacking ornithine-delta-aminotransferase have paradoxical neonatal hypoornithinaemia and retinal degeneration. Nat Genet. 1995 Oct;11(2):185-90.
[5]Boffa I, Polishchuk E, De Stefano L, Dell'Aquila F, Nusco E, Marrocco E, Audano M, Pedretti S, Caterino M, Bellezza I, Ruoppolo M, Mitro N, Cellini B, Auricchio A, Brunetti-Pierri N. Liver-directed gene therapy for ornithine aminotransferase deficiency. EMBO Mol Med. 2023 Apr 11;15(4):e17033.
[6]Geng R, Omar A, Gopal SR, Chen DH, Stepanyan R, Basch ML, Dinculescu A, Furness DN, Saperstein D, Hauswirth W, Lustig LR, Alagramam KN. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci Rep. 2017 Oct 18;7(1):13480.
[7]Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003 Dec 1;12(23):3075-86.
[8]Tian G, Lee R, Ropelewski P, Imanishi Y. Impairment of Vision in a Mouse Model of Usher Syndrome Type III. Invest Ophthalmol Vis Sci. 2016 Mar;57(3):866-75.
[9]Geng R, Omar A, Gopal SR, Chen DH, Stepanyan R, Basch ML, Dinculescu A, Furness DN, Saperstein D, Hauswirth W, Lustig LR, Alagramam KN. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci Rep. 2017 Oct 18;7(1):13480.
[10] Geng R, Geller SF, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, Jones SM, Wright CG, Melki S, Imanishi Y, Palczewski K, Alagramam KN, Flannery JG. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum Mol Genet. 2009 Aug 1;18(15):2748-60.
[11] Dinculescu A, Stupay RM, Deng WT, Dyka FM, Min SH, Boye SL, Chiodo VA, Abrahan CE, Zhu P, Li Q, Strettoi E, Novelli E, Nagel-Wolfrum K, Wolfrum U, Smith WC, Hauswirth WW. AAV-Mediated Clarin-1 Expression in the Mouse Retina: Implications for USH3A Gene Therapy. PLoS One. 2016 Feb 16;11(2):e0148874.
[12] Brambati M, Borrelli E, Sacconi R, Bandello F, Querques G. Choroideremia: Update On Clinical Features And Emerging Treatments. Clin Ophthalmol. 2019 Nov 18;13:2225-2231.
[13] Kalatzis V, Roux AF, Meunier I. Molecular Therapy for Choroideremia: Pre-clinical and Clinical Progress to Date. Mol Diagn Ther. 2021 Nov;25(6):661-675.
[14] Tolmachova T, Anders R, Abrink M, Bugeon L, Dallman MJ, Futter CE, Ramalho JS, Tonagel F, Tanimoto N, Seeliger MW, Huxley C, Seabra MC. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest. 2006 Feb;116(2):386-94.
[15] Tolmachova T, Wavre-Shapton ST, Barnard AR, MacLaren RE, Futter CE, Seabra MC. Retinal pigment epithelium defects accelerate photoreceptor degeneration in cell type-specific knockout mouse models of choroideremia. Invest Ophthalmol Vis Sci. 2010 Oct;51(10):4913-20.
[16] Cehajic Kapetanovic J, Barnard AR, MacLaren RE. Molecular Therapies for Choroideremia. Genes (Basel). 2019 Sep 23;10(10):738.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



