

WGS(Whole Genome Sequencing)의 발전에 따라, intronic 돌연변이가 다양한 질환, 특히 희귀 질환에서 중요한 역할을 한다는 사실이 점점 더 많이 인식되고 있습니다. 정상적인 유전자 전사(gene transcription) 과정에서, exon과 intron 모두 전구체 메신저 RNA(pre-mRNA)로 복사된 후, splicing 과정에서 intron이 제거되어 최종 단백질로 번역될 수 있는 Coding Sequence(CDS)가 형성됩니다.
연구에 따르면, 약 10%~30%의 질환 관련 유전자 돌연변이(주로 intronic 돌연변이)는 splicing에 영향을 미치거나 enhancer나 silencer와 같은 전사 조절 인자를 방해하여, 잠재적인 splice site 활성화, Pseudoexon Inclusion, Exon Skipping 등의 메커니즘을 유발하여 궁극적으로 질환을 일으킵니다. 이러한 메커니즘은 종종 PTC(premature termination codon)를 생성하여 NMD(Nonsense-mediated RNA Decay), 단백질 2차 구조 변형 또는 유전자/단백질 발현 수준의 불규칙성을 일으킵니다. [1-3]
본문은 intronic 돌연변이로 인해 발생하는 질환의 대표적인 사례 -- 가족성 자율신경 실조증(FD, Familial Dysautonomia)에 대해 다룬 인간화 유전자 연구 모델을 소개합니다.
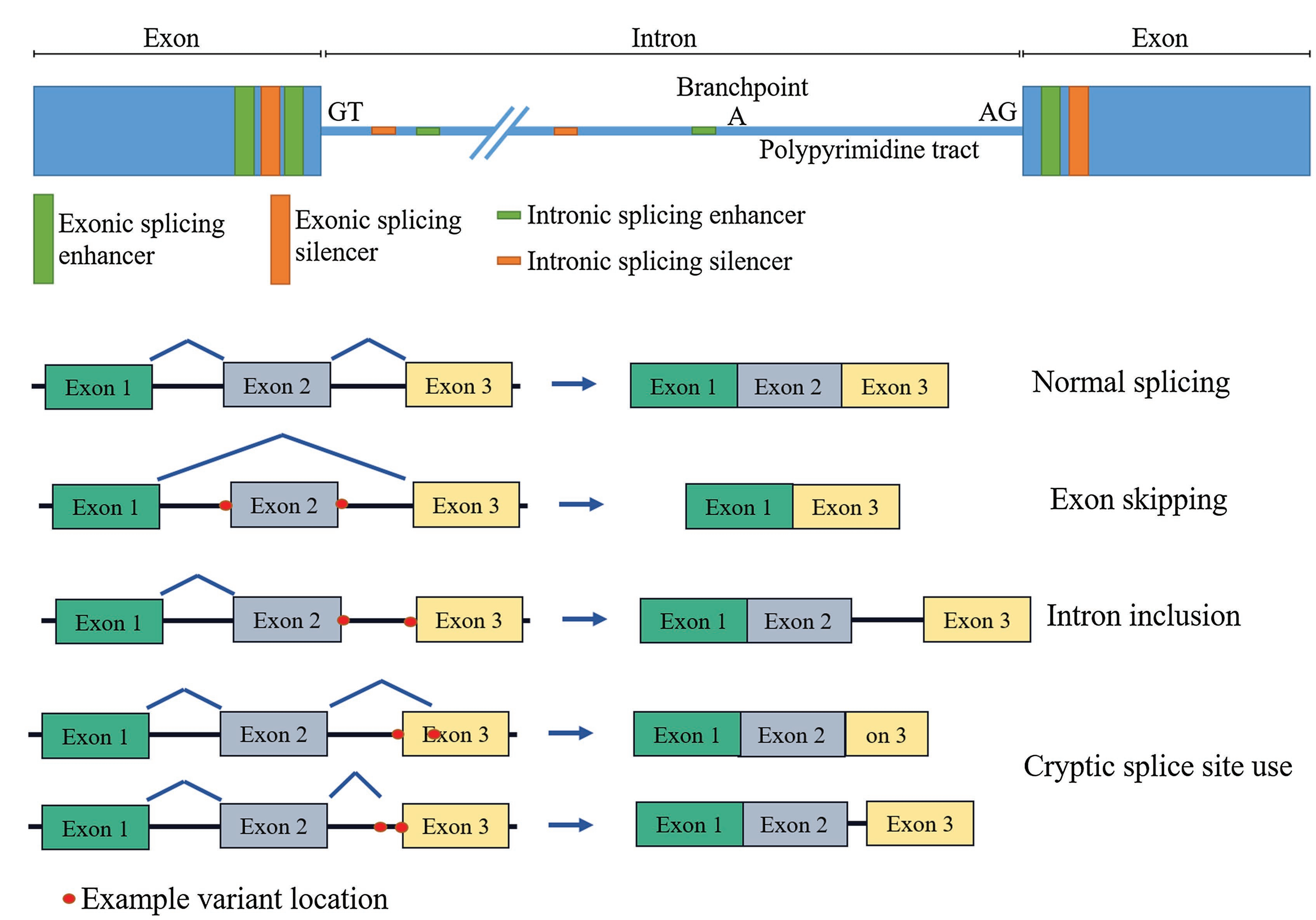
그림 1. intronic 돌연변이가 pre-mRNA splicing에 미치는 일반적인 영향. [3]
가족성 자율신경 실조증(FD) 및 ELP 유전자 intronic 돌연변이
가족성 자율신경 실조증(FD)은 신경 세포 발달 장애와 중추 신경계 퇴행으로 인해 발생하는 희귀 유전성 신경 장애입니다. 자율 신경계(Autonomic nervous system)와 감각 신경계(Sensory nervous system)의 결함으로 인해, 환자는 과도한 발한, 간헐적인 고혈압, 침 흘림, 연하곤란/삼킴장애, 불규칙한 장 및 방광 조절, 호흡 곤란, 주기적인 구토 등의 증상이 나타납니다.
이 질환은 특정 민족 및 인종 집단과 관련되어 있으며, 주로 아슈케나지 유대인(Ashkenazi Jews)에게 영향을 미치며, 이 인종 집단에서 발생률은 약 3,600명 중 1명입니다. 환자의 약 50%만이 40세까지 생존합니다. [4-5]
ELP1(IKBKAP) 유전자는 신경 세포의 발달과 기능에 중요한 역할을 하는 elongator complex의 구성 요소를 암호화합니다. 대부분의 가족성 자율신경 실조증(FD) 환자는 ELP1 유전자에서 이중대립유전자성 돌연변이(Biallelic mutation)를 보이며, 그중 99% 이상의 환자는 intron 20(IVS20+6T>C)의 5' splice site에서 돌연변이가 발생합니다. 이러한 돌연변이는 U1 snRNP (Small nuclear ribonucleoprotein)와 intron 20의 donor splice site 사이의 염기쌍(Base pairing)을 방해하여 exon 20의 skipping을 초래합니다. [4-6] 이 잘못된 splicing은 transcript reading frame에서 프레임시프트(frameshift)를 일으켜 PTC(Premature Termination Codon )를 생성하고, 이에 따라 잘린 ELP 단백질이 합성되어 결국 신경 세포 손상과 세포 사멸을 초래합니다.
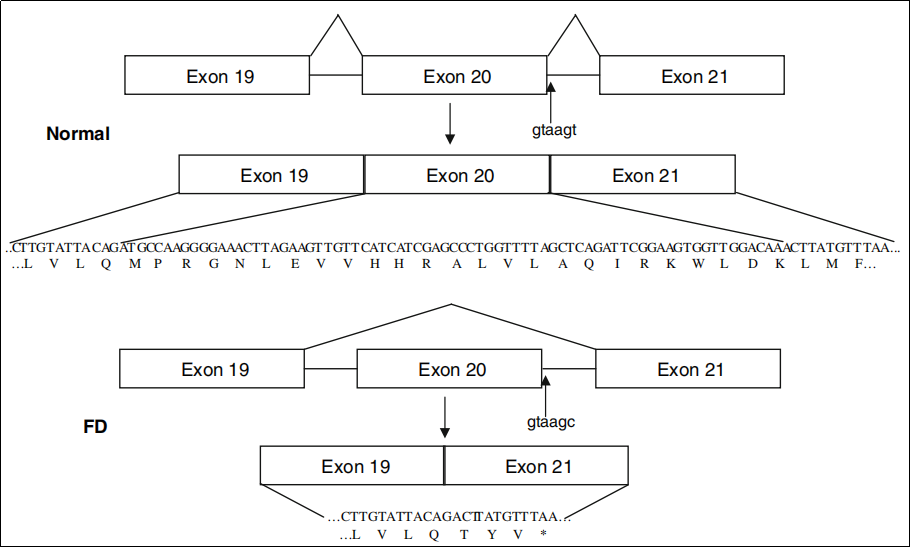
그림 2. ELP1/IKBKAP intronic 돌연변이 IVS20+6T>C가 비정상적인 transcript splicing을 일으키는 메커니즘 [6]
가족성 자율신경 실조증(FD): ELP1을 타겟팅하는 치료법 및 관련 동물 모델
현재 가족성 자율신경 실조증(FD)에 대한 치료법은 없으며, 치료 전략은 주로 대증치료 및 보조 치료에 초점을 맞추어 증상을 완화하고 합병증을 예방하는 데 집중하고 있습니다.
IVS20+6T>C 돌연변이는 가족성 자율신경 실조증(FD)에서 가장 흔한 병리적 돌연변이로, 이 돌연변이에 의해 발생하는 splicing 오류를 수정하여 전장 ELP1 단백질을 생성하는 연구가 진행되고 있습니다. 미국 Cold Spring Harbor Laboratory와 PTC Therapeutics의 연구자들은 이 분야에서 안티센스 올리고뉴클레오티드(ASO) 및 저분자 약물 개발을 포함한 중요 연구를 수행하였습니다. [7-9]
연구에 따르면, wild-type(wt) 마우스를 사용하여 ELP1 유전자 intronic 돌연변이의 splicing 패턴을 조사하는 것은 효과적이지 않습니다. 그 이유는 Elp1 homozygous knockout 마우스가 배아 발달 단계에 사망하기 때문입니다. 인간 변이 ELP1 유전자를 발현하는 transgenic 마우스는 내인성 Elp1 유전자 정상 수준의 발현으로 인해 명확한 질환 표현형을 나타내지 않습니다. 따라서 이 방법은 마우스 내인성 Elp1 유전자에서 Elp1 유전자 knockdown (single-copy knockout) 접근법과 transgenic 유전자를 결합해야 합니다. 그러나 이 접근법은 여전히 불안정한 transgene copy와 일관되지 않은 표현형 등 문제에 직면해 있습니다. [10-12] 또한, 마우스와 인간 간의 splicing 패턴 차이로 인해 마우스 Elp1 유전자의 exon 20을 인간화하여 그 flanking intron과 함께 IVS20+6T>C 돌연변이를 도입하는 것도 표현형을 생성하는 데 실패했습니다. [13]
이러한 연구들은 마우스에서 ELP1 유전자의 splicing 패턴을 조사하려면 더 길거나 심지어 전장 인간 ELP1 유전자 염기서열이 필요한한 것을 시사합니다.
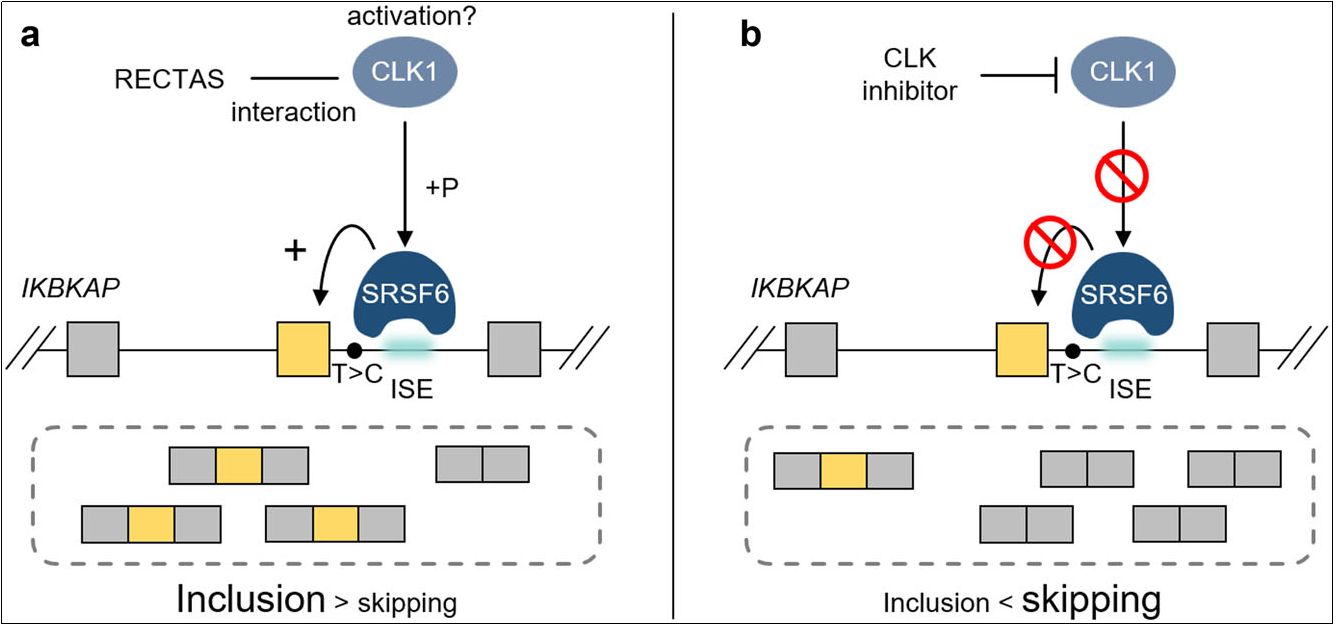
그림3. 저분자 약물을 통해 IKBKAP IVS20+6T>C의 exon 20 splicing 패턴을 조절합니다. [9]
효과적인 가족성 자율신경 실조증(FD) 연구에 대한 요구를 충족시키기 위해, Cyagen은 마우스 Elp1 유전자의 서열(개시 코돈에서 종결 코돈까지)을 해당 인간 ELP1 유전자 서열로 in situ 대체한B6-hELP1 인간화 마우스 모델(제품 번호: I001203)을 개발하였습니다. 또한, 가족성 자율신경 실조증(FD) 연구를 지원하기 위해, Cyagen은 B6-hELP1 모델을 바탕으로 IVS20+6T>C 인간화 가족성 자율신경 실조증(FD) Point Mutation 모델을 개발하고 있습니다.
인간 ELP1 유전자를 성공적으로 발현하는 B6-hELP1 마우스
유전자 발현 분석 결과, B6-hELP1 마우스의 대뇌피질, 신장, 간, 골격근, 심장 등 여러 조직에서 인간 ELP1 유전자의 유의미한 발현이 검출되며, 마우스 내인성 Elp1 유전자 mRNA 발현이 검출되지 않았습니다.
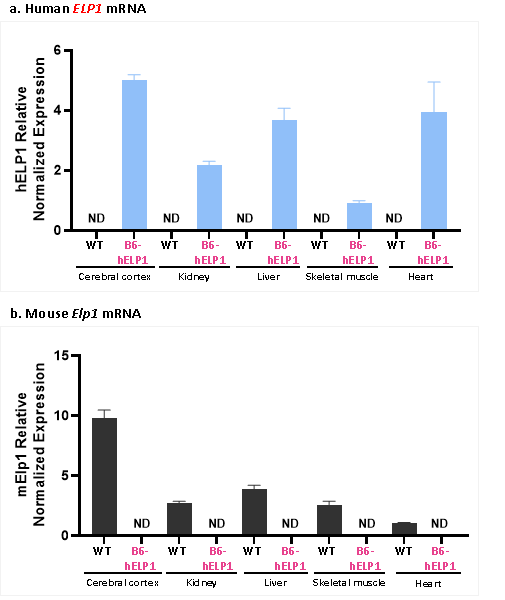
그림 4. B6-hELP1 마우스와 wild-type(wt) 마우스에서 인간 ELP1 유전자와 마우스 내인성 Elp1 유전자의 발현 결과.
요약
가족성 자율신경 실조증(FD) 치료에 관한 기존 연구는 주로 돌연변이에 의한 splicing 오류를 교정하여 전장 ELP1 단백질을 생성하는 데 집중하고 있습니다. B6-hELP1 모델 (제품 번호: I001203)은 내인성 마우스 Elp1 유전자의 간섭 없이 마우스에서 전장 인간 ELP1 유전자를 발현시켜 가족성 자율신경 실조증(FD) 연구에 적용됩니다. 예비 연구에 따르면, B6-hELP1 마우스를 기반으로 구축한 IVS20+6T>C 인간화 Point Mutation 모델(개발 중)은 인간 가족성 자율신경 실조증(FD) 환자에서 나타나는 표현형과 유사한 표현형을 보일 것으로 예상됩니다.
B6-hELP1 모델 외에도, Cyagen은 신경학적, 안과적 및 기타 질환을 위한 다양한 유전자 인간화 모델을 개발하여 여러 질환에 대한 타겟 치료제를 개발하는 연구자들에게 강력한 지원을 제공합니다.
|
제품 번호 |
모델명 |
품종 계통 |
응용 분야 |
|
C001396 |
C57BL/6JCya |
Retinitis Pigmentosa (RP), Congenital Stationary Night Blindness (CSNB), and other retinal diseases. |
|
|
C001410 |
C57BL/6JCya |
Frontotemporal Dementia (FTD), Alzheimer's Disease (AD), and other neurodegenerative diseases. |
|
|
C001418 |
C57BL/6JCya |
Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Dementia (FTD), and other neurodegenerative diseases. |
|
|
C001427 |
C57BL/6NCya |
Parkinson's Disease (PD). |
|
|
C001437 |
C57BL/6NCya |
Spinal Muscular Atrophy with Respiratory Distress Type 1 (SMARD1) and Charcot-Marie-Tooth Disease Type 2S (CMT2S). |
|
|
C001495 |
C57BL/6JCya |
Retinitis pigmentosa (RP), congenital stationary night blindness (CSNB), and other retinal diseases research |
|
|
C001504 |
C57BL/6NCya |
Spinal muscular atrophy (SMA) |
|
|
I001128 |
C57BL/6NCya |
Rett Syndrome (RTT) |
|
|
I001124 |
C57BL/6NCya |
Hutchinson-Gilford Progeria Syndrome (HGPS) |
|
|
C001398 |
C57BL/6NCya |
Spinocerebellar Ataxia Type 3 (SCA3) |
|
|
C001512 |
C57BL/6NCya |
Transthyretin Amyloidosis (ATTR) |
|
|
I001131 |
C57BL/6NCya |
Epilepsy |
|
|
I001132 |
C57BL/6NCya |
Cystic Fibrosis (CF) |
|
|
C001525 |
C57BL/6NCya |
Transthyretin Amyloidosis (ATTR) |
|
|
I001130 |
C57BL/6NCya |
Hepatolenticular Degeneration (HLD) |
|
|
IR1019 |
Sprague-Dawley |
Alexander disease (AxD), traumatic brain injury |
|
|
C001533 |
C57BL/6NCya |
Obesity, metabolic disorders associated with improper fat distribution and storage |
|
|
C001538 |
C57BL/6NCya |
Dystrophic Epidermolysis Bullosa (DEB) |
|
|
C001428 |
C57BL/6NCya |
Epidermolysis Bullosa (EB) |
|
|
C001546 |
C57BL/6JCya |
Corneal Dystrophy (CD) |
|
|
C001551 |
C57BL/6JCya |
Stargardt Disease (STGD) |
|
|
C001554 |
C57BL/6JCya |
Usher Syndrome (USH) |
|
|
C001555 |
C57BL/6JCya |
Age-related Macular Degeneration (AMD); Diabetic Retinopathy (DR); Corneal Neovascularization; Mechanisms of Tumorigenesis and Development, and Development of Antitumor Drugs. |
|
|
I001191 |
C57BL/6JCya |
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Lobar Degeneration/Dementia (FTLD) |
|
|
I001181 |
C57BL/6JCya |
Frontotemporal Dementia (FTD), Alzheimer's Disease (AD), and other neurodegenerative diseases. |
|
|
I001182 |
C57BL/6JCya |
Frontotemporal Dementia (FTD), Alzheimer's Disease (AD), and other neurodegenerative diseases. |
참고 문헌:
[1]Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019 Jan 24;176(3):535-548.e24.
[2]Chiang HL, Chen YT, Su JY, Lin HN, Yu CA, Hung YJ, Wang YL, Huang YT, Lin CL. Mechanism and modeling of human disease-associated near-exon intronic variants that perturb RNA splicing. Nat Struct Mol Biol. 2022 Nov;29(11):1043-1055.
[3]Lord J, Baralle D. Splicing in the Diagnosis of Rare Disease: Advances and Challenges. Front Genet. 2021 Jul 1;12:689892.
[4]Rubin BY, Anderson SL. IKBKAP/ELP1 gene mutations: mechanisms of familial dysautonomia and gene-targeting therapies. Appl Clin Genet. 2017 Dec 15;10:95-103.
[5]Dietrich P, Dragatsis I. Familial Dysautonomia: Mechanisms and Models. Genet Mol Biol. 2016 Oct-Dec;39(4):497-514. doi: 10.1590/1678-4685-GMB-2015-0335. Epub 2016 Aug 4.
[6]Rubin BY, Anderson SL. The molecular basis of familial dysautonomia: overview, new discoveries and implications for directed therapies. Neuromolecular Med. 2008;10(3):148-56.
[7]Morini E, Gao D, Montgomery CM, Salani M, Mazzasette C, Krussig TA, Swain B, Dietrich P, Narasimhan J, Gabbeta V, Dakka A, Hedrick J, Zhao X, Weetall M, Naryshkin NA, Wojtkiewicz GG, Ko CP, Talkowski ME, Dragatsis I, Slaugenhaupt SA. ELP1 Splicing Correction Reverses Proprioceptive Sensory Loss in Familial Dysautonomia. Am J Hum Genet. 2019 Apr 4;104(4):638-650.
[8]Sinha R, Kim YJ, Nomakuchi T, Sahashi K, Hua Y, Rigo F, Bennett CF, Krainer AR. Antisense oligonucleotides correct the familial dysautonomia splicing defect in IKBKAP transgenic mice. Nucleic Acids Res. 2018 Jun 1;46(10):4833-4844.
[9]Ajiro M, Awaya T, Kim YJ, Iida K, Denawa M, Tanaka N, Kurosawa R, Matsushima S, Shibata S, Sakamoto T, Studer L, Krainer AR, Hagiwara M. Therapeutic manipulation of IKBKAP mis-splicing with a small molecule to cure familial dysautonomia. Nat Commun. 2021 Jul 23;12(1):4507.
[10]Dietrich P, Yue J, E S, Dragatsis I. Deletion of exon 20 of the Familial Dysautonomia gene Ikbkap in mice causes developmental delay, cardiovascular defects, and early embryonic lethality. PLoS One. 2011;6(10):e27015.
[11]Hims MM, Shetty RS, Pickel J, Mull J, Leyne M, Liu L, Gusella JF, Slaugenhaupt SA. A humanized IKBKAP transgenic mouse models a tissue-specific human splicing defect. Genomics. 2007 Sep;90(3):389-96.
[12]Morini E, Dietrich P, Salani M, Downs HM, Wojtkiewicz GR, Alli S, Brenner A, Nilbratt M, LeClair JW, Oaklander AL, Slaugenhaupt SA, Dragatsis I. Sensory and autonomic deficits in a new humanized mouse model of familial dysautonomia. Hum Mol Genet. 2016 Mar 15;25(6):1116-28.
[13]Bochner R, Ziv Y, Zeevi D, Donyo M, Abraham L, Ashery-Padan R, Ast G. Phosphatidylserine increases IKBKAP levels in a humanized knock-in IKBKAP mouse model. Hum Mol Genet. 2013 Jul 15;22(14):2785-94.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



