

낭포성 섬유증(Cystic fibrosis, CF)은 전 세계 수만 명의 환자에게 영향을 미치는 치명적인 진행성 유전 질환으로, 현재까지 치료법이 없습니다. 낭포성 섬유증(CF) 환자의 85% 이상은 F508del 변이를 가지고 있기 때문에 정확하고 신뢰할 수 있는 전임상 연구 모델을 개발할 필요성이 날로 커지고 있습니다. Cyagen에서 최신 개발한 B6-hCFTR*F508del 인간화 마우스 모델은 연구의 새로운 가능성을 열어줍니다. — 낭포성 섬유증(CF) 병리학을 정확하게 재현하여 약물 발견과 치료법 혁신을 가속합니다.
낭포성 섬유증(CF) 소개
낭포성 섬유증(CF)은 생명을 위협하는 진행성 유전 질환으로, 주로 백인 Caucasian에 영향을 미칩니다. 이 질환은 CFTR(Cystic Fibrosis Transmembrane Conductance Regulator) 유전자의 돌연변이로 인해 점액은 비정상적으로 두꺼워져 호흡기 및 소화기 계통을 심각하게 손상시킵니다. 낭포성 섬유증(CF)은 비정상적으로 두꺼워진 점액이 기도와 췌관을 막아 호흡 곤란, 재발성 폐 감염, 췌장 기능 부전, 영양실조 등의 증상으로 로 이어져 환자의 삶의 질을 크게 떨어뜨립니다.
치료법의 발전으로 환자의 기대 수명이 연장되었지만, 낭포성 섬유증(CF)은 여전히 완치가 불가능하고 장기적인 치료와 관리가 필요한 만성 질환입니다. 따라서 낭포성 섬유증(CF)을 완치하기 위해서는 보다 효과적인 치료법을 개발하는 것이 중요합니다. 낭포성 섬유증(CF) 환자의 85% 이상이 F508del 유전자의 돌연변이를 가지고 있어 치료제 개발의 핵심 타겟이 되고 있습니다.
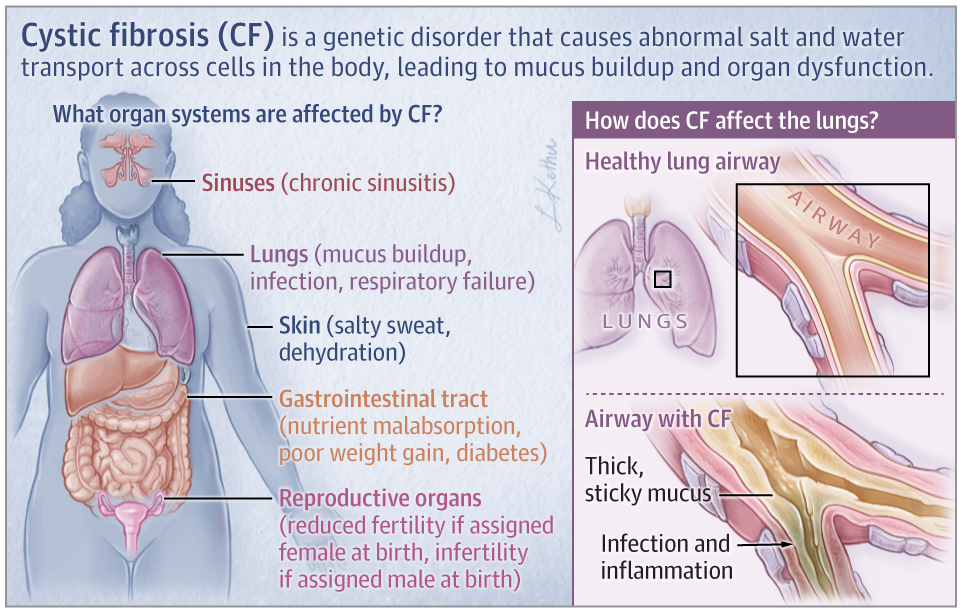
그림 1. 낭포성 섬유증(CF)의 주요 증상.[1]
CFTR(Cystic Fibrosis Transmembrane Conductance Regulator): 중요한 이온 채널(Ion Channel) 단백질
CFTR(Cystic Fibrosis Transmembrane Conductance Regulator)은 폐, 췌장, 땀샘 등 다양한 신체 기관의 상피 조직에서 염분과 수분 균형을 유지하는 데 중요한 역할을 하는 중요한 transmembrane 이온 채널(Ion Channel) 단백질입니다. CFTR의 주요 기능은 염화 이온(Chloride ion) 채널로서 상피 세포막을 통한 염화 이온 및 중탄산 이온(Bicarbonate ion)의 수송을 조절하여 조직액 균형과 pH를 유지하는 것입니다. 이 과정은 ATP 가수분해에 의존하며 다른 이온 채널과 수송 단백질의 활성을 조절할 수 있습니다[2].
F508del 변이를 포함한 CFTR 유전자의 돌연변이는 염화 이온 채널 기능 이상을 초래하여 낭포성 섬유증(CF)을 비롯한 다양한 질환을 유발합니다.
낭포성 섬유증(CF)은 특히 백인 Caucasian 사이에서 많이 발생하고 출생아 2,500명에서 1,800명당 1명의 비율로 발병하며, 전 세계 환자 수는 약 9만 명으로 추정됩니다[2]. 낭포성 섬유증(CF)의 전형적인 표현형 특성으로는 비정상적으로 두꺼운 폐의 점액, 빈번한 호흡기 감염, 췌장 기능 부전, 남성 불임(일반적으로 정관 폐쇄와 연관됨) 등이 있습니다.
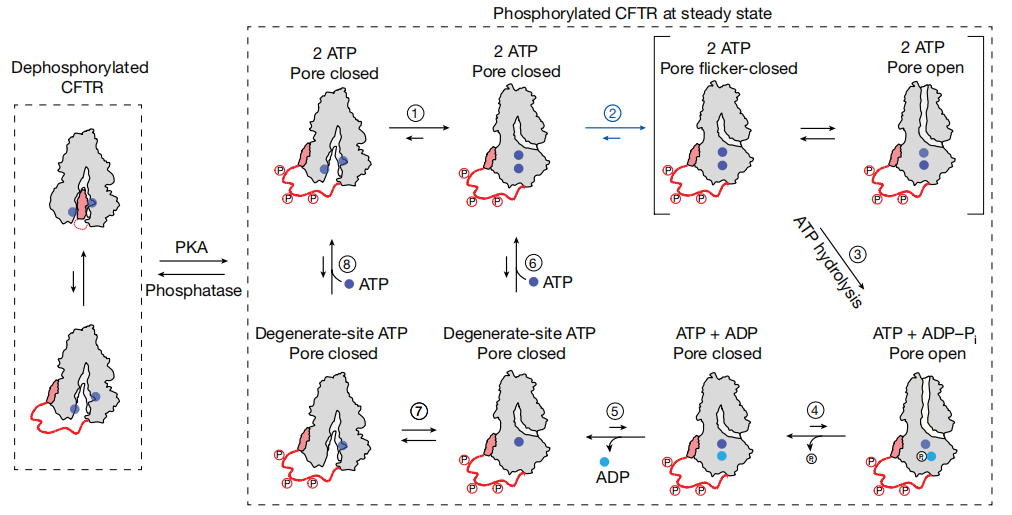
그림 2. CFTR gated channel의 형태 변화는 생리적 및 약리학적 조절의 핵심입니다. [3]
F508del (ΔF508) 병원성 돌연변이
F508del-CFTR로도 알려진 F508del(c.1521_1523delCTT)은 낭포성 섬유증(CF)에서 가장 흔한 병원성 돌연변이로, 환자의 85% 이상이 이 변이의 대립 유전자를 최소 하나로 가지고 있으며, 그 중 약 40%는 homozygous(동형 접합)입니다[4-5].
CFTR 단백질은 생합성 및 점힘 과정에서 복잡한 도메인 조립을 거치는데, F508del 돌연변이는 CFTR 단백질의 첫 번째 뉴클레오타이드 결합 도메인(NBD1)에서 아미노산 페닐알라닌(F508)의 결실을 초래하여 잘못된 단백질 점힘을 일으킵니다. 새로 합성된 CFTR 단백질은 소포체를 떠나기 전에 소포체 관련 단백질 분해(ERAD) 경로를 분류되고, 유비퀴틴-프로테아좀 시스템(Ubiquitin-Proteasome System, UPS)에 의해 분해됩니다.
이로 인해 염화 이온 채널 기능에 이상이 발생하여 만성 폐쇄성 폐질환(Chronic Obstructive Pulmonary Disease, COPD)을 유발합니다.[6] 그 결과 F508del은 낭포성 섬유증(CF) 치료제 개발의 주요 타겟 중 하나가 되었습니다.
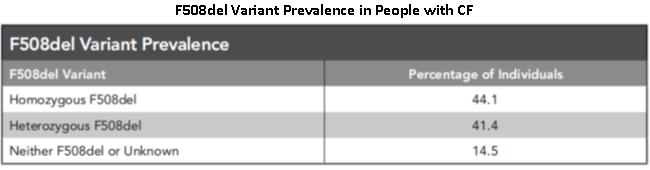
그림 3. 낭포성 섬유증(CF) 환자의 85% 이상은 F508del 돌연변이 유전자의 copy를 최소 하나로 보유하고 있습니다. [5]
예를 들어, Vertex Pharma에서 개발한 CFTR 조절제는 F508del-CFTR 돌연변이 단백질의 형태 안정성을 향상시키고 분해를 줄이며, 세포 표면에서 CFTR 단백질의 발현과 기능을 개선하는 효능이 입증되었습니다. 이러한 CFTR 단백질 기능 개선은 환자의 질환 표현형 완화에 도움이 됩니다.[7-8] 그러나 기존 치료법의 한계를 극복하고 치료법을 개선하기 위해서는 지속적인 연구가 필요합니다.
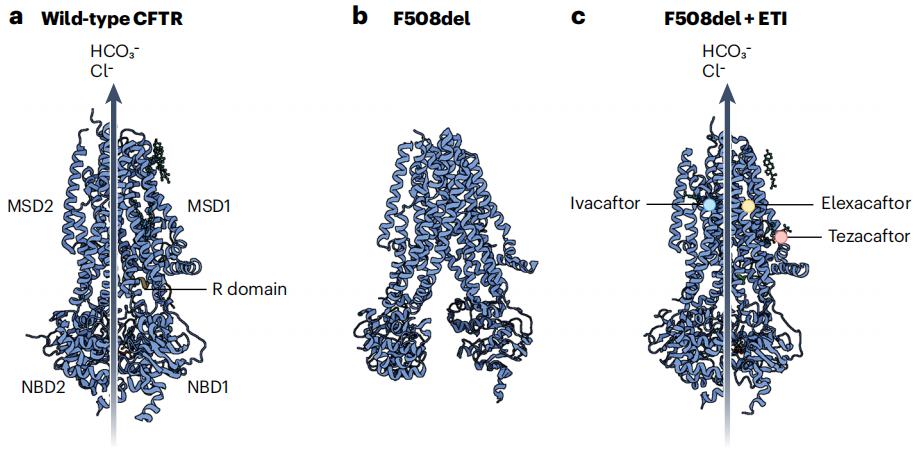
그림 4. Wild-type(WT) CFTR 단백질과 F508del-CFTR 돌연변이 단백질의 구조 및 CFTR 조절제의 작용 메커니즘. [9]
Cyagen의 최신 인간화 CFTR 및 인간화 Point Mutation 모델
인간화 Point Mutation 마우스 모델은 낭포성 섬유증(CF) 타겟팅 치료법에 대한 전임상 in vivo 평가의 정확성을 크게 향상할 수 있습니다. Cyagen은 낭포성 섬유증(CF) 연구를 발전시키기 위해 B6-hCFTR 및 B6-hCFTR*F508del 인간화 마우스 모델을 개발하였습니다. 이러한 마우스 모델은 낭포성 섬유증(CF) 관련 질환 특성을 나타내며, 낭포성 섬유증(CF)의 발병 기전을 연구하고 CFTRF508del 돌연변이를 타겟으로 하는 치료법을 평가할 수 있는 유용한 연구 모델입니다.
주요 특징:
연구에 따르면 B6-hCFTR 및 B6-hCFTR*F508del 모델 모두 간, 장, 폐를 포함한 주요 조직에서 인간화된 CFTR mRNA를 발현하며 마우스 내인성 Cftr 유전자 발현이 없는 것으로 확인되었습니다.
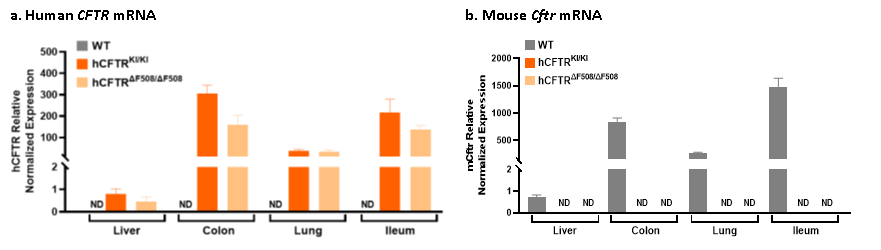
그림 5. CFTR 인간화 마우스 모델의 in vivo 유전자 발현
CFTR 단백질 발현 수준
B6-hCFTR과 B6-hCFTR*F508del 마우스 모두 인간 CFTR 단백질을 성공적으로 발현하였습니다. 특히, B6-hCFTR*F508del 마우스의 CFTR 발현 수준이 B6-hCFTR 마우스보다 낮은 것으로 검증되며, 이는 F508del 돌연변이의 병리학적인 영향을 반영합니다.
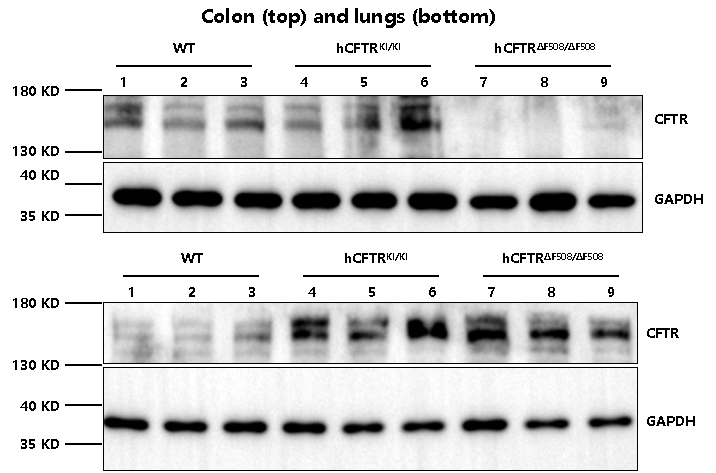
그림 6. 마우스 모델의 in vivo CFTR 단백질 발현 비교
B6-hCFTR*F508del 마우스는 배상세포(Goblet cell) 증식, 점액 축적, 장벽 두께 증가 등 낭포성 섬유증(CF)의 대표적인 병리학적 특징을 보였습니다. 반면, B6-hCFTR 마우스는 경미한 조직 병리학적 변화만 보였고, Wild-type(WT) 마우스는 정상적인 조직을 보였습니다. 이러한 표현형은 낭포성 섬유증(CF) 발병 기전과 치료 접근법을 연구하는 데 있어 B6-hCFTR*F508del 마우스 모델의 유용성을 입증합니다.
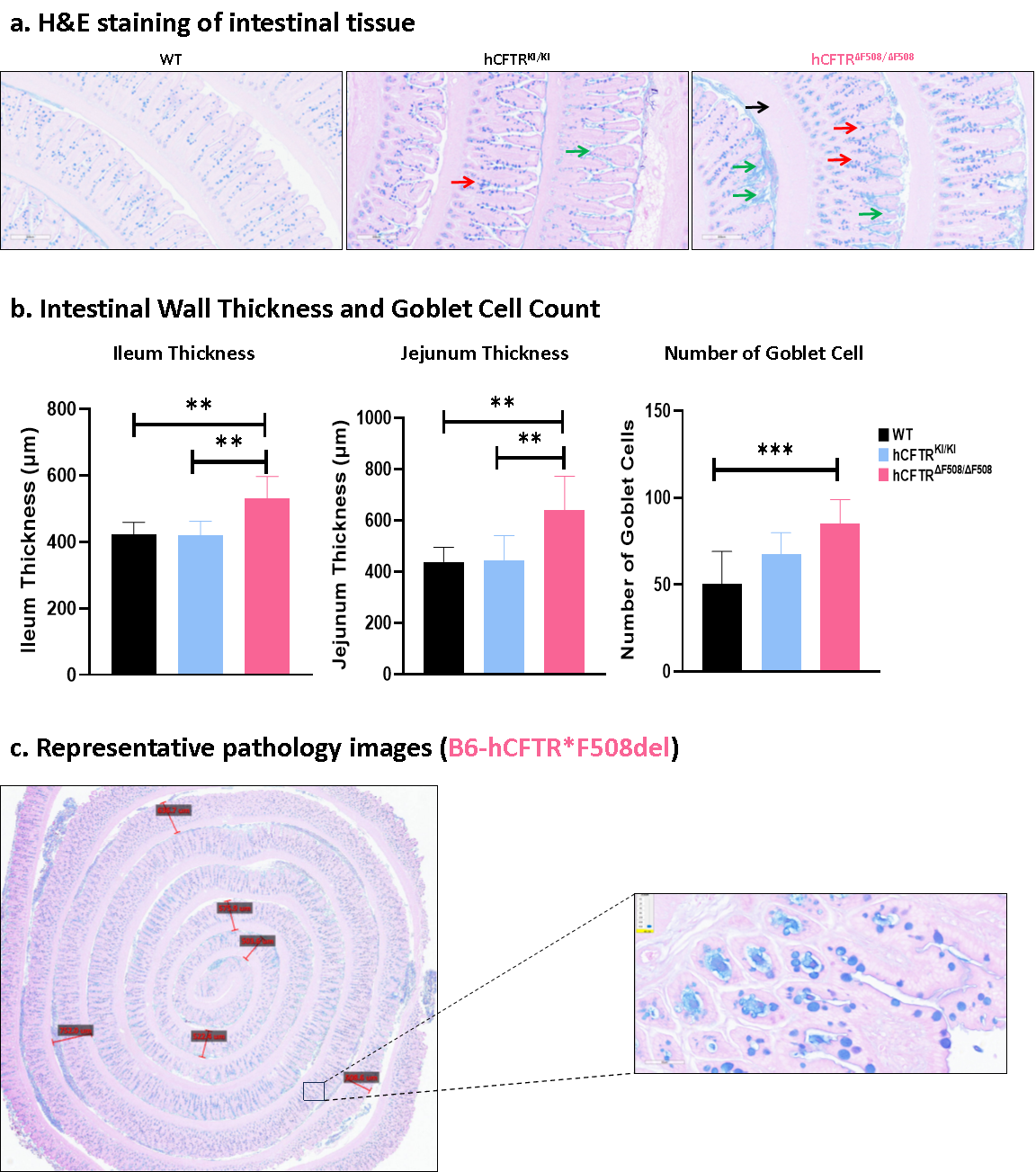
그림 7. 마우스 모델의 장 조직 병리학적 특징 비교
요약
B6-hCFTR 마우스(제품 번호: I001132)와 B6-hCFTRF508del 마우스(제품 번호: I001226)는 마우스 내인성 Cftr 유전자의 발현 없이 인간 CFTR 유전자 및 단백질을 성공적으로 발현합니다. 낭포성 섬유증(CF) 연구를 위한 이러한 인간화 마우스 모델의 주목할 만한 장점은 다음과 같습니다:
상기 연구 결과에 의하면, B6-hCFTR 마우스와 B6-hCFTRF508del 마우스 모두 인간 CFTR 유전자 및 단백질 발현에 상당한 이점을 가지고 있으며 인간 낭포성 섬유증(CF)의 특징을 효율적으로 시뮬레이션할 수 있습니다.
이러한 모델은 다음과 같은 연구 분야에 적용됩니다:
Cyagen은 또한 신경학, 대사, 희귀 질환 등 다양한 연구 분야의 과학자들의 연구 니즈를 충족하기 위해 완전 인간화 모델과 인간화 Point Mutation 질환 모델을 개발하였습니다.
참고 문헌:
[1]Endres TM, Konstan MW. What Is Cystic Fibrosis? JAMA. 2022;327(2):191.
[2]Grasemann H, Ratjen F. Cystic Fibrosis. N Engl J Med. 2023 Nov 2;389(18):1693-1707.
[3]Levring J, Terry DS, Kilic Z, Fitzgerald G, Blanchard SC, Chen J. CFTR function, pathology and pharmacology at single-molecule resolution. Nature. 2023 Apr;616(7957):606-614.
[4]Ong T, Ramsey BW. Cystic Fibrosis: A Review. JAMA. 2023 Jun 6;329(21):1859-1871.
[5]Cystic Fibrosis Foundation. (2021). 2021 Patient Registry Annual Data Report. Retrieved December 12, 2024, from https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf
[6]McDonald EF, Woods H, Smith ST, Kim M, Schoeder CT, Plate L, Meiler J. Structural Comparative Modeling of Multi-Domain F508del CFTR. Biomolecules. 2022 Mar 18;12(3):471.
[7]Carnovale V, Scialò F, Gelzo M, Iacotucci P, Amato F, Zarrilli F, Celardo A, Castaldo G, Corso G. Cystic Fibrosis Patients with F508del/Minimal Function Genotype: Laboratory and Nutritional Evaluations after One Year of Elexacaftor/Tezacaftor/Ivacaftor Treatment. J Clin Med. 2022 Nov 22;11(23):6900.
[8]Riepe C, Wąchalska M, Deol KK, Amaya AK, Porteus MH, Olzmann JA, Kopito RR. Small-molecule correctors divert CFTR-F508del from ERAD by stabilizing sequential folding states. Mol Biol Cell. 2024 Feb 1;35(2):ar15.
[9]Mall MA, Burgel PR, Castellani C, Davies JC, Salathe M, Taylor-Cousar JL. Cystic fibrosis. Nat Rev Dis Primers. 2024 Aug 8;10(1):53.



영업일 기준 1-2일 내에 답변해 드리겠습니다.



