

Note: This research proposal is developed by Lindsay Marjoram, PhD on behalf of the Barth Syndrome Foundation.
Research Proposal
1. Summary of the Proposed Study
Background: Barth syndrome (BTHS; OMIM #302060) is a life-limiting, X-linked ultra-rare disorder predominantly affecting males. It is caused by mutations in the transacylase TAFAZZIN, which prevents production of mature cardiolipin1,2 (CL) and leads to impaired cellular energetics. Individuals with BTHS suffer from cardio- and skeletal myopathy, neutropenia, failure to thrive and profound and crushing fatigue3,4. These symptoms lead to consequences such as fatal arrhythmias, frequent infections, the need for implantable cardiac devices and/or heart transplants5. Previous research from patient samples and yeast models has demonstrated that mutations in TAFAZZIN and accumulation of an immature CL called mono-lyso cardiolipin (MLCL) lead to defects in mitochondrial organization and subsequently impaired energy production.
Creating a viable animal model for BTHS posed significant challenges until recently in part due to failure to transmit the tafazzin allele through the germline7. The first viable mouse model of BTHS was sponsored by the Barth Syndrome Foundation and generated by Taconic Labs. Using a doxycycline-inducible, shRNA-mediated approach, Tafazzin can be knocked down and this model recapitulated many of the hallmarks characteristic of BTHS6. However, chronic administration of doxycycline and differences in knockdown approaches can lead to significant variability in this model7,8. Additionally, there is still residual functional Tafazzin which is not typically the case in humans7. A conditional Tafazzin knockout mouse line was generated and characterized8. This model exhibits phenotypes resembling human Barth syndrome, including cardiomyopathy and skeletal muscle weakness as well as perinatal lethality8. Most recently, a knock-in (KI) tafazzin allele corresponding to a known patient mutation was generated in mice9. Similar to the conditional knockout, the KI mouse line recapitulates many aspects of BTHS. In the conditional knockout and KI lines, male mice are sterile, which is not the case in humans7. While gene therapy has shown promise in correcting these deficits, the scalability and durability of the treatment remain obstacles for human applications. Moreover, it is unclear if regulatory regions (e.g. intronic sequences) in the human TAFAZZIN gene contribute to the phenotypes observed in affected individuals.
Biological Questions:
Several biological questions remain to be answered that, to date, cannot be evaluated in human iPSC or current mouse models:
·Does a fully-genomically humanized mouse model recapitulate the full spectrum of Barth Syndrome phenotypes observed in humans?
·Are there functional differences between a humanized mouse model for the tafazzin locus and the conditional knockout and knock-in models that already exist?
·What are the effects of pathologic tafazzin gene mutations on whole-body physiology and lipid metabolism in a fully-genomically humanized mouse model and do these differ from the mouse models previously generated?
·Can novel therapies targeting TAFAZZIN and other affected pathways be optimized and tested in these models for better translatability to human treatments?
·Would this mouse model represent a high throughput platform for evaluating variants of unknown significance that have been identified in the BTHS community?
Significance: The goal of this study is to develop a fully-genomically humanized mouse model of BTHS using HUGO-GT™ technology. This model will enable a deeper understanding of the disease’s molecular mechanisms and facilitate the development and efficacy evaluation of potential therapies. The expected outcomes have the potential to significantly impact the field of genetic and rare/ultra-rare disorders, particularly in developing novel treatments for BTHS.
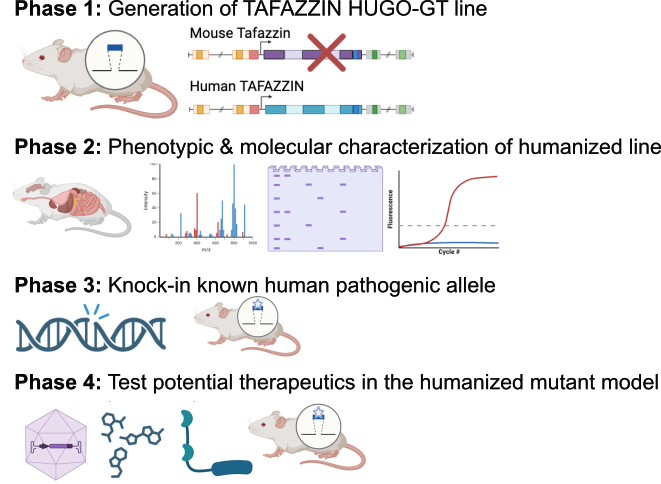
2. Description of the HUGO-GT Model(s)
Model Construction: The proposed model involves the creation of a fully-genomically humanized mouse model using HUGO-GT™ technology, which ensures full-length coverage of disease hotspot mutation regions, including exons, introns, and untranslated regions (UTRs). This differs from traditional models, which only achieve partial human gene insertion. The Cyagen TurboKnockout® and BAC fusion technologies will be employed to replace large-scale genomic segments, enabling precise and efficient creation of the humanized model.
Technical Feasibility:
Cyagen has already validated these novel genetic approaches, which reduces the technical risks associated with making a new mouse model.
·TurboKnockout® Technology: Allows for rapid and efficient replacement of large genomic segments.
·BAC Fusion Technology: Facilitates the insertion of full-length human gene sequences, ensuring accurate representation of the human TAFAZZIN gene.
Experimental Plan:
·Phase 1: Develop the fully-genomically humanized mouse model by replacing the mouse tafazzin gene with the human equivalent, including all regulatory elements.
·Phase 2: Characterize the physiological, biochemical, and molecular phenotypes of the humanized mice, focusing on heart and skeletal muscle tissues.
·Phase 3: Generate known pathologic allele knock-in mice on the HUGO-GT™ tafazzin background to evaluate mutations within the different functional domains of the Tafazzin protein.
·Phase 4: Test the efficacy of novel therapies including gene therapy targeting the TAFAZZIN gene in the humanized mouse model. Assess the therapeutic outcomes in terms of gene uptake efficiency, long-term expression, and functional correction of disease phenotypes.
3. Expected Outcomes and Impact
Anticipated Outcomes:
·Development of a robust and accurate fully-genomically humanized mouse model of Barth Syndrome.
·Comprehensive understanding of the effects of TAFAZZIN gene mutations on whole-body physiology.
·Identification of potential therapeutic targets and optimization of gene therapy strategies for Barth Syndrome.
Potential Impact:
·Translational Potential: The fully-genomically humanized model offers a greater degree of translatability from preclinical studies to clinical applications, addressing the current limitations of existing models and sparing the use of larger animal models, which will yield significant cost savings.
·Therapeutic Development: The insights gained from this model could facilitate the identification and development of the novel therapy classes for Barth Syndrome, significantly improving patient outcomes.
·Broader Implications: The methodologies and findings from this study could be applied to other genetic disorders, enhancing the overall field of gene therapy and precision medicine.
This proposal aims to leverage the advanced capabilities of HUGO-GT™ technology to create a novel and highly relevant animal model of BTHS, providing a critical tool for advancing our understanding and treatment of this life-limiting disease.
참고문헌: